|
Читайте также: |
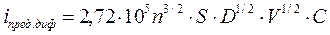 , (43)
, (43)
где V- скорость изменения поляризующего напряжения.
Переменнотоковая вольтампорометрия. В классическом способе вольтамперометрии на электроды ячейки подается постоянный ток с изменяющимся во времени напряжением. В данном же способе анализа на электроды наряду с постоянным напряжением, медленно изменяющимся во времени, накладывается переменное напряжение небольшой амплитуды (до 50 мВ). Это дает возможность уменьшить почти до нуля конденсаторный ток, который сильно искажает форму вольтамперных.
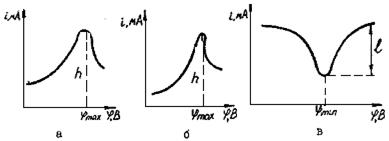
Рис.34. Вольтамперные кривые:
А - с линейной разверткой напряжения; б - переменнотоковая;
в - инверсионная кривых при малых концентрациях вещества в растворе.
Вследствие чего увеличивается чувствительность и разрешающая способность данного способа анализа.
Так же, как и в вольтамперометрии с линейной разверткой, вольтамперная переменнотоковая кривая имеет форму кривой с максимумом (рис.34,б) и содержит такую же аналитическую информацию: потенциал максимума характеризует природу вещества, а максимальная высота пропорциональна его концентрации.
Инверсионная вольтамперометрия. Определяемое вещество с концентрацией 10-7-10-9 м/л некоторое время подвергают электролизу в небольшом объеме стационарного ртутного электрода или на поверхности твердого электрода при потенциале, несколько более отрицательном, чем потенциал полуволны определяемого иона. Определяемое вещество, например, в случае полярографии, при этом концентрируется в ртути в виде амальгамы. Последняя затем анодно растворяется при потенциале, непрерывно изменяющемся от значения, при котором проводилось катодное выделение элемента на ртути, до более положительных потенциалов. В результате кривая, анодного тока имеет вид характерного зубца (рис.34, в), глубина l которого соответствует iпред.диф и пропорциональна концентрации определяемого иона в растворе при условии постоянства других факторов. Значение φmin характеризует природу вещества.
2.2.5. Аппаратура. Для проведения вольтамперометрического анализа необходимы электролитическая ячейка и прибор-полярограф. Электроли-тическая ячейка состоит из стеклянного сосуда емкостью от 1 до 50 мл с погруженными в него рабочим минроэлектродом и вспомогательным электродом. Микроэлектродом могут служить либо ртутная капля, либо такие металлы, как Рt, Au, Ag и т.п., а также графит специальной обработки. В качестве вспомогательного электрода используют слой ртути с большой поверхностью либо электроды сравнения (каломельный, хлорсеребряный и т.п.).
Полярограф представляет собой прибор, поляризующий электроды ячейки постоянно изменяющимся напряжением и в тоже время регистрирующий изменение силы тока в ячейке от подаваемого напряжения.
В ходе анализа вольтамперная кривая записывается пером на диаграммной бумажной ленте, которая перемещается синхронно с подаваемым напряжением. В результате получается вольтамперная кривая в координатах «i – φ». Если скорость изменения напряжения, подаваемого на ячейку, велика (до нескольких десятков вольт в секунду), самопишущие регистраторы в силу их инерционности использовать нельзя. Тогда вольтамперная кривая регистрируется на экране осциллографа, и определение iпред.диф и 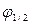 производят по осциллограммам.
производят по осциллограммам.
В настоящее время выпускают различные марки полярографов: ППТ-1, ПУ-1, IP-7, I П-7в, ОН-102, OH-105 и др.
В заключение следует отметить, что помимо анализа растворов, содержащих ионы неорганических веществ, вольтамперометрия дает неплохие результаты при анализе многих органических веществ. К последним относятся соединения, содержащие карбонильные группы, двойные углерод-углеродные связи, связи углерод-галоген, азот-кислород, диазогруппы. Соединения такого типа характерны для процессов и продуктов бумажной, гидролизной и лесохимической промышленности, а также для производства древесных плит и пластиков. Поскольку многие органические соединения плохо растворяются в воде, в качестве растворителей часто используют органические жидкости.
2.3. Амперометрическое титрование. При проведении амперометрического титрования необходимо соблюдать условия вольтамперометрического анализа:
- один из электродов ячейки должен иметь очень малую поверхность (индикаторный микроэлектрод), поверхность другого электрода должна быть велика (вспомогательный электрод). В этом случае поляризуется только индикаторный микроэлектрод, потенциал вспомогательного электрода остается практически постоянным;
- в исследуемый раствор необходимо вводить фоновый электролит (в результате чего падением напряжения в электролите можно пренебречь);
- ионы определяемого вещества должны восстанавливаться или окисляться на индикаторном микроэлектроде.
Дополнительным условием для проведения амперометрического титрования является то, что оно проводится при потенциале предельного тока диффузии. Изменение последнего в процессе титрования раствора является аналитическим сигналом в этом методе анализа. Действительно, в соответствии с уравнением Ильковича (37) 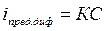 величина предельного тока диффузии пропорциональна концентрации вещества в растворе. Таким образом, изменение концентрации определяемого иона повлечет за собой изменение величины iпред.диф (рис.35,а).
величина предельного тока диффузии пропорциональна концентрации вещества в растворе. Таким образом, изменение концентрации определяемого иона повлечет за собой изменение величины iпред.диф (рис.35,а).
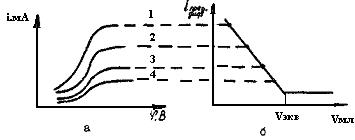
Рис.35. Вольтамперные кривые при различной концентрации
вещества в растворе (а) и кривая амперометрического титрования (б); С1>С2>С3>С4
Если по оси ординат отложить значения iпред.диф, а по оси абсцисс - объем добавляемого титранта, то в результате получим кривую амперометрического титрования (рис.35,б).
Кривая амперометрического титрования состоит из двух прямолинейных участков, пересечение которых соответствует точке эквивалентности.
В зависимости от характера реакции, протекающей между определяемым веществом и титрантом, а также от того, какое вещество окисляется или восстанавливается на индикаторном микроэлектроде при потенциале титрования, различают несколько типов кривых амперометрического титрования.
Первый тип кривой (рис.36,а) наблюдается, когда исследуемое вещество электрохимически активно, т.е. при заданном потенциале окисляется или восстанавливается на индикаторном микроэлектроде. Титрант в этом случае индифферентен (не участвует в электродных реакциях).
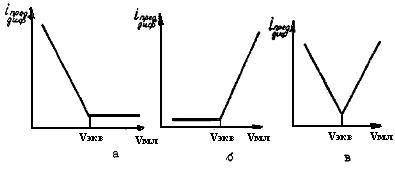
Рис.36. Типы кривых амперометрического титрования
Кривые первого типа получаются при титровании ионов Ag+, Рb2+, Bi3+ или каких-либо других ионов, способных восстанавливаться на индикаторном микрокатоде, каким-либо осадителем.
Например, раствор AgN03 титруется раствором КI при потенциале предельного тока диффузии разряда ионов Ag+ на катоде Ag+ + е → Ag. В результате реакции осаждения Ag+ + I- → AgI концентрации ионов Ag+ в исследуемом растворе будет уменьшаться. Следовательно, в соответствии c уравнением Илъковича (37), для данного случая по мере добавления титранта будет уменьшаться значение предельного тока диффузии.
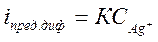
В точке эквивалентности предельный ток диффузии достигает минимального значения и далее остается постоянным.
Второй тип кривых наблюдается тогда, когда ионы исследуемого вещества при заданном потенциале электрохимически неактивны (не участвуют в электродной реакции), а титранта электрохимически активен. Например, раствор, содержащий ионы Ва2+, титруется хроматом:
Ba2+ + Cr2-4 = BaCrO4
До точки эквивалентности (рис.36,б) диффузионный ток иона CrO4 невелик (соответствует остаточному току полярографической волны), т.к. концентрация ионов CrO42- в растворе мала. По мере добавления титранта концентрация ионов СrО42-достигает такой величины, что становится возможным их разряд на рабочем микроэлектроде:
CrO2-4 + 3e +8H+ Þ Cr3+ + 4H2O
Дальнейшее добавление титранта в соответствии с уравнением Ильковича 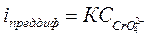 повлечет за собой увеличение предельного тока диффузии. Третий тип кривых наблюдается, когда оба компонента - ионы исследуемого вещества и титрант - принимают участие в электрохимических реакциях. Например, раствор, содержащий ионы Рb2+, титруется хроматом:
повлечет за собой увеличение предельного тока диффузии. Третий тип кривых наблюдается, когда оба компонента - ионы исследуемого вещества и титрант - принимают участие в электрохимических реакциях. Например, раствор, содержащий ионы Рb2+, титруется хроматом:
Pb2+ + CrO2-4 Þ PbCrO4
Вначале при добавлении титранта снижение 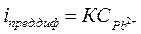 обусловливается уменьшением концентрации ионов Pb2+, связывающихся в осадок. В точке эквивалентности ток имеет минимальное значение. При дальнейшем добавлении титранта в растворе увеличивается концентрация ионов СrO42-, что влечет за собой возрастание (рис.36, в).
обусловливается уменьшением концентрации ионов Pb2+, связывающихся в осадок. В точке эквивалентности ток имеет минимальное значение. При дальнейшем добавлении титранта в растворе увеличивается концентрация ионов СrO42-, что влечет за собой возрастание (рис.36, в).
В разобранных примерах использовался один индикаторный микроэлектрод. Если определяемый ион восстанавливается, микроэлектрод должен быть катодом, если окисляется - анодом.
Однако возможно амперометрическое титрование с двумя индикаторными электродами. В этом случае в анализируемый раствор помещают два одинаковых электрода, между которыми поддерживается небольшая разность потенциалов (10-50 мВ). Наличие тока в ячейке связано с электрохимическими процессами на двух электродах. При титровании по этому методу часто отпадает необходимость в построении кривой титрования, т.к. точка эквивалентности может быть определена по резкому прекращению или появлению тока.
В процессе амперометрического титрования происходит увеличение объема раствора. Чтобы не вводить поправку на изменение объема, концентрация раствора титранта должна быть в 10-20 раз выше концентрации исследуемого вещества.
2.3.1. Аппаратура. Установка для амперометрического титрования компактна и собирается из доступных и недорогих приборов (рис.37).
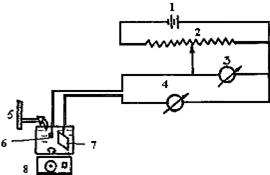
Рис.37. Амперометрическая установка с одним индикаторным микроэлектродом
В нее входят: источник постоянного тока 1 (аккумулятор, сухой элемент), потенциометр, или магазин переменного сопротивления 2 (примерно на 1 кОм), вольтметр постоянного тока 3, микроамперметр постоянного тока 4 чувствительностью 10-6-10-9 А, микробюретка 5, электролитическая ячейка с индикаторным микроэлектродом 6 и вспомогательным электродом 7, магнитная мешалка 8.
Установив потенциометром 2 на вольтметре 3 значение потенциала предельного тока диффузии определяемого иона, в ячейку из микробюретки 5 добавляют титрант и следят за показаниями микроамперметра 4. Строят график в координатах «iпред.диф -V мл» и определяют эквивалентный объем титранта, пошедшего на титрование. Рассчитывают концентрацию исследуемого вещества в анализируемом растворе по формуле:
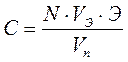 , (44)
, (44)
где
С - концентрация исследуемого вещества, г/л;
N - нормальность стандартного раствора титранта, экв/л;
Vэ - объем стандартного раствора титранта, пошедшего на титрование, мл;
Э -эквивалент исследуемого вещества;
Vn - объем пробы исследуемого вещества, мл.
В качестве индикаторного микроэлектрода применяют ртутный капающий или твердый электрод (Pt, Au, Ti, W, графит и т.д.). В качестве вспомогательного электрода используют слой ртути на дне ячейки или металл (последний должен быть нерастворим); можно использовать также каломельный или хлорсеребряный электроды сравнения.
При титровании с двумя индикаторными электродами используют два одинаковых поляризуемых электрода (например, платиновых) площадью 1-2 см2 каждый.
Аналитические возможности метода амперометрического титрования широки. Этим методом можно определять практически все элементы периодической системы и большое число органических соединений, используя реакции осаждения, комплексообразования, окисления - восстановления и кислотно-основного взаимодействия. Нижний предел определяемых концентраций 10-6 м/л. Метод прост и не требует сложной аппаратуры.
2.4. Кулонометрия. Кулонометрия объединяет методы анализа, основанные на измерении количества электричества, затраченного на электрохимическую реакцию определяемого иона. Аналитическим сигналом в данном методе анализа является сила тока, проходящего через электролитическую ячейку в процессе электролиза.
2.4.1. Электролиз. Процесс электролиза проводят в электрохимической ванне, являющейся системой, в которой за счет приложенного извне постоянного электрического тока происходят химические превращения веществ на электродах (см.рис.1). Таким образом, электролизом называются химические превращения веществ под действием электрического тока. Для процесса электролиза необходимо, чтобы на электродах ванны были достигнуты потенциалы разряда (выделения) ионов, находящихся в растворе электролита. Тогда на катоде ванны пойдут процессы восстановления, например:
Men+ +n е → Me0,
на аноде - процессы окисления:
Ме0 –n е→ Men+
Основные законы электролиза установлены Фарадеем:
- количество вещества, выделившееся на электродах при электролизе, пропорционально количеству электричества, прошедшему через раствор электролита;
- при прохождении через раствор электролита одного и того же количества электричества на электродах выделяется одно и тоже количество эквивалентов вещества. Эти законы выражаются формулой:
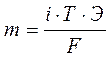 , (45)
, (45)
где
m - масса вещества, выделившегося на электродах, г;
i - сила тока, прошедшего через электролит, А;
Т - время, с;
Э - г-эктз вещества;
F - число, или постоянная, Фарадея, 96496 Кл.
Законы Фарадея являются одними из наиболее точных законов природа. Согласно им, при прохождении через растворы электролитов количества электричества, равного одному Фарадею, на электродах должен выделиться 1 г-экв вещества: 1 г водорода; 8 г кислорода; 63,5 г меди; 107,8 г серебра и т.д.
Однако в практической деятельности часто встречаются видимые отклонения от законов Фарадея. Например, при электролизе ZnCl2на катоде выделяется цинк: Zn2++2e→Zn°
При прохождении через раствор количества электричества, равного одному Фарадею, на катоде должен выделиться 1г-экв цинка (32,7 г). Однако в действительности цинка на катоде выделяется меньше. Это обусловлено тем, что на катоде, помимо выделения цинка, может идти выделение водорода из молекул воды: 2Н2О + 2е → H2f + 2OH- или разряд растворенного в воде кислорода: O2 + 2H2O + 4e → 4OH.
Поскольку на осуществление этих реакций тратится какое-то количество электричества, на процесс выделения цинка его остается меньше чем один Фарадей. В результате цинка на катоде выделяется меньше одного грамм-эквивалента.
Подобные отклонения от законов Фарадея обусловлены совместным разрядом ионов. Они могут возникнуть и тогда, когда продукты электролиза, выделившиеся на электродах, частично растворяются; могут быть и другие случаи.
Обычно в процессах электролиза используется величина, называемая «выход по току», которая рассчитывается по формуле:
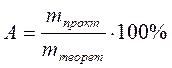 , (46)
, (46)
где
А - выход по току, %;
mпракт - масса вещества, выделившегося на электроде в процессе электролиза, г;
mтеорет - масса вещества, которая должна выделиться на электроде теоретически в соответствии с формулой (45), г.
Если отклонений от закона Фарадея нет, масса вещества, выделившегося на электроде, равна массе вещества, рассчитанной по формуле (45), и выход по току составит 100%. Если в процессе электролиза существуют видимые отклонения от законов Фарадея, то mпрокт будет меньше mтеорет и выход по току будет меньше 100%.
Из законов Фарадея следует, что количество электричества, прошедшее через раствор электролита, может быть при известных условиях определено по массе продуктов электролиза, выделившихся на электроде при прохождении тока. Прошедшее количество электричества измеряют с помощью кулонометров.
Кулонометр представляет собой электрохимическую ванну, в которой протекает хорошо известная электрохимическая реакция со 100%-м выходом по току. Принцип действия кулонометра основан на том, что его включают в электрическую цепь последовательно с электрохимической ванной, в которую залит анализируемый раствор. Таким образом, за некоторый промежуток времени через анализируемый раствор в ванне и кулонометр пройдет одно и то же количество электричества. Поскольку в кулонометре проходит известная электрохимическая реакция (например, Сu2+ + 2 е → Сu0), то измерение количества электричества сводится к определению массы вещества, выделившегося на катоде кулонометра.
Действительно, зная массу выделившейся на катоде меди и ее грамм-эквивалент, по формуле (45) можно рассчитать количество электричества, прошедшего через анализируемый раствор:
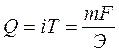 . (47)
. (47)
В зависимости от способа измерения различают электрогравиме-трические, газовые, титрационные и электронные кулонометры. В электрогравиметрическах кулонометрах определяют массу, например, металлической меди, выделившейся при электролизе сульфата меди. В газовых кулонометрах определяется объем газа, выделившегося в результате электрохимического процесса, и по этому объему рассчитывают количество электричества. Электронный кулонометр непосредственно отсчитывает число кулонов.
2.4.2. Теоретические основы метода. Кулонометрия основана на законах Фарадея, дающих связь между количеством электричества, прошедшего через раствор электролита, и массой вещества, выделившегося на электроде в результате электрохимической реакции. Если известны количество электричества и природа вещества, можно определить весовое содержание, или концентрацию определяемого вещества в растворе. При этом необходимо, чтобы все расходуемое количество электричества затрачивалось на электрохимическую реакцию определяемого вещества, т.е. чтобы процесс протекал со 100%-м выходом по току.
В зависимости от происходящих в растворе электрохимических процессов различают прямую кулонометрию и косвенную (кулонометрическое титрование).
В свою очередь, прямая кулонометрия подразделяется на потенциостатическую кулонометрию и амперостатическую кулонометрию). В первом случае анализ выполняется при поддержании постоянного значения потенциала рабочего электрода, во втором - при постоянной величине тока.
Потенциостатическая кулонометрия. Каждый ион, участвующий в электрохимической реакции, выделится на электроде только в том случае, если достигнет его потенциал разряда. В противном случае электрохимического акта не будет.
На рис.38 представлена вольтамперная кривая для многокомпонентной системы.
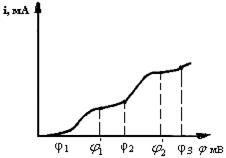
Рис.38. Вольтамперная кривая многокомпонентной системы
При значении потенциала электрода φ1 начинается разряд одного из компонентов раствора, потенциал φ2 соответствует началу разряда второго компонента. При потенциале, φ 3 начинает разряжаться третий компонент. Таким образом, устанавливая то или иное значение потенциала электрода, можно добиться, чтобы на рабочем электроде выделялся определенный ион. Например, если нам необходимо проанализировать первый компонент, то значение потенциала рабочего электрода не должно превышать φ1. При этом условии на рабочем электроде будет выделяться только первый компонент системы, т.к. потенциалы выделения второго и третьего компонентов не достигнуты. Следует иметь в виду, однако, что если достигнут потенциал φ3, то на рабочем электроде будет выделяться не только третий компонент, но также первый и второй. Это необходимо учитывать при проведении данного вида анализа.
Потенциостатическая кулонометрия основана на измерении количества электричества, прошедшего на электрохимическое восстановление или окисление определяемого вещества при постоянном значении потенциала рабочего электрода. Чтобы процесс шел с максимальной скоростью и 100%-м выходом по току, электролиз следует проводить при потенциале, соответствующем середине площадки предельного тока диффузии определяемого вещества. Для первого компонента этот потенциал соответствует φ1 (см. рис.38).
По мере восстановления или окисления на электродах определяемого иона его концентрация в растворе электролита уменьшается и сила тока в цепи падает. Общее количество электричества, затраченное на полное электрохимическое превращение определяемого иона, выражается площадью, ограниченной кривой в координатах «сила тока – время» (рис.39,а) и рассчитывается по формуле:
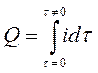 . (48)
. (48)
До полного электрохимического превращения исследуемого вещества нужно бесконечно большое время, т.к. iτ → 0 при τ → ∞. Поэтому для практических целей величину тока в любой момент времени можно определить по уравнению:
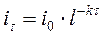 , (49)
, (49)
где
iτ - величина тока к моменту времени t, А;
i0 - ток в момент начала электролиза, А;
К - константа, зависящая от условий электролиза, с-1.
В соответствии с уравнением (49), уменьшение тока идет по экспоненциальной кривой (рис.39,б).
Прологарифмировав уравнение (49), получим:
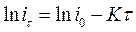 . (50)
. (50)
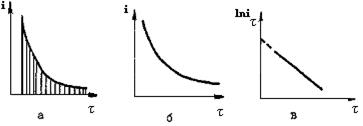
Рис.39. Зависимости силы тока от времени электролиза:
а- теоретическая -кривая ток-время в потенциостатических условиях;
б- кинетическая кривая в потенциостатических условиях;
в- кинетическая кривая в полулогарифмических координатах
Полученное выражение представляет собой прямую линию (рис.39,в), точка пересечения которой с осью ординат соответствует 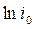 , а тангенс угла ее наклона - К. Определив значение К количество электричества находят по формуле:
, а тангенс угла ее наклона - К. Определив значение К количество электричества находят по формуле:
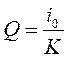 . (51)
. (51)
Рассмотренный расчетный способ определения количества электричества предложен Мак-Невилом и Байкером. Кроме того, количество электричества, необходимое для проведения анализа, может быть также определено с помощью химического или электронного кулонометров.
Содержание определяемого вещества находят по закону Фарадея. Простейшая потенциостатическая схема представлена на рис.40:
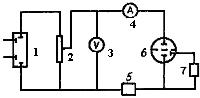
Рис.40. Схема установки для потенциостатической кулонометрии:
1- источник постоянного тока; 2- делитель напряжения; 3- вольтметр;
4- миллиамперметр; 5- кулонометр; 6- электрохимическая ячейка с пористой перегородкой, разделяющей катодное и анодное пространства;
7- блок измерения электродного потенциала
Для того, чтобы на электродах поддерживалась постоянная разность потенциалов, сопротивление ячейки 6 должно быть во много раз меньше сопротивления делителя напряжения 2. Для устранения влияния продуктов электролиза, образующихся на вспомогательном электроде, катодное и анодное пространства должны быть разделены пористой перегородкой или электролитическим ключом. Об окончании процесса электролиза можно судить по резному уменьшению тока (практически до нуля), регистрируемого миллиамперметром 4.
Преимуществом потенциостатический кулонометрии является высокая селективность метода т.е. возможность поочередного определения отдельных веществ в их смеси.
Амперостатическая кулонометрия. Основана на определении количества электричества, затраченного на электрохимические превращения определяемого вещества при постоянном значении тока, протекающего через электролитическую ячейку.
Поскольку определение содержащегося в растворе вещества при этом методе проводят при постоянном значении силы тока, то, определив время электролиза, можно рассчитать количество электричества по формуле:
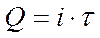 . (52)
. (52)
Однако в практике проведения этого вида анализа дело обстоит сложнее. В процессе электролиза по мере уменьшения концентрации анализируемого вещества изменяется потенциал рабочего электрода и появляется возможность разряда другого компонента раствора. Следовательно, будут происходить затраты электричества на побочные электрохимические процессы, что скажется на точности результатов анализа (выход по току для определяемого вещества будет меньше 100%). Чтобы избежать этого, принимаются специальные меры. Например, в исследуемый раствор вводится вспомогательный реагент, который участвует в электрохимической реакции, а продукт этой реакции должен стехиометрически взаимодействовать с определяемым веществом. Простейшая гальваностатическая схема представлена на рис.41.
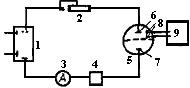
Рис.41. Схема установки для амперостатической кулонометрии:
1- источник постоянного тока; 2- высокоомное сопротивление;
3- миллиамперметр; 4- кулонометр; 5- электрохимическая ячейка с пористой перегородкой, разделяющей катодное и анодное пространства;
6- рабочий электрод; 7- вспомогательный электрод; 8- индикаторные электроды; 9- индикаторный блок
Для определения момента окончания электролиза применяют как визуальные (с помощью индикаторов), так и инструментальные (потенциометрия, амперометрия, фотометрия и др.) методы. Иногда об окончании процесса можно судить по изменению потенциала электрода.
Кулонометрическое титрование. Если в прямой кулонометрии определяемое вещество подвергается непосредственному электрохимическому превращению, то в кулонометрическом титровании определяемое вещество А может быть неэлектроактивно. В электрохимическую реакцию на рабочем электроде вступает вспомогательный реагент В, добавляемый в ячейку, который, например, восстанавливается до промежуточного реагента С:
В + е → С.
Затем в растворе происходит химическая реакция А + С → АС. Таким образом, кулонометрическое титрование основано на электрохимическом получении промежуточного реагента, стехиометрически вступающего в химическую реакцию с определяемым веществом.
В качестве химической реакции, протекающей между определяемым веществом и промежуточным реагентом, может быть использована любая реакция, применяемая в титриметрии.
Электролиз ведут при постоянной силе тока, а расчет количества электричества, израсходованного на генерацию дополнительного реагента (до промежуточного реагента) и, следовательно, на превращение определяемого вещества, производят по формуле (33). Для того, чтобы изменение концентрации вспомогательного реагента в процессе электролиза было незначительным, его обычно берут в 1000-кратном избытке по отношении в определяемому веществу. При этом обеспечивается стопроцентный выход по току.
Схема установки для кулонометрического титрования такая же, как и для амперостатической кулонометрии (см. рис.41). Окончание, процесса электролиза определяют так же, как в амперостатической кулонометрии.
Дата добавления: 2015-07-21; просмотров: 164 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА. 3 страница | | | ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА. 5 страница |