Читайте также:
|
| Приёмы количественного определения в хроматографии |
В качестве аналитического сигнала в хроматографии используют высоту или площадь хроматографического пика, которые пропорциональны содержанию вещества в хроматографической зоне.
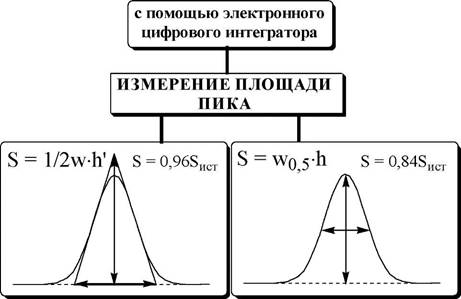
|
Приёмы количественного определения, используемые в хрома- тографических методах, приведены в табл. 22.3.
Табл. 22.3.
|
Внутренний стандарт представляет собой специально добавляемое к анализируемой пробе в точно измеренном количестве вещество, свойства которого близки к свойствам определяемого вещества. Вещество, взятое в качестве внутреннего стандарта:
• не должно химически взаимодействовать с компонентами анализируемой смеси, с неподвижной или подвижной фазой;
• должно отсутствовать в исходной анализируемой смеси;
• давать пик, находящийся на хроматограмме в непосредственной близости от пиков определяемых веществ, но не накладывающийся на них и на пики других соединений.
Концентрацию внутреннего стандарта выбирают таким образом, чтобы высота (площадь) пика внутреннего стандарта была соизмерима с высотой (площадью) пика определяемого вещества (рис. 22.3).
растворитель
В методе внешнего и внутреннего стандарта используются одни и те же приёмы расчёта содержания вещества (метод градуировочного графика и др.). Основное различие заключается в характере используемого аналитического сигнала. Метод внутреннего стандарта обладает большей надёжностью и даёт более воспроизводимые результаты, особенно в случае сложной пробоподготовки.
22.4. Теории хроматографического разделения
 Рис. 22.3. Хроматограмма, полученная при ГЖХ-определении аминазина в печени (внутренний стандарт - бромгексин)
Рис. 22.3. Хроматограмма, полученная при ГЖХ-определении аминазина в печени (внутренний стандарт - бромгексин)
|
При хроматографировании происходят два процесса: разделение веществ и размывание хроматографических зон разделяемых веществ. Хроматографический процесс заключается в многократном повторении актов сорбции и десорбции. Поскольку скорость сорбции и де
сорбции для молекул различных веществ различна, то после повторения большого числа элементарных актов хроматографического разделения при прохождении смеси веществ через слой сорбента происходит разделение её на отдельные компоненты. Положение и вид хрома- тографических зон разделяемых веществ зависят от формы изотермы сорбции, скорости установления равновесия, степени диффузии вещества в подвижной фазе.
Изотермой сорбции называется зависимость концентрации вещества, сорбированного неподвижной фазой, от его концентрации в подвижной фазе при постоянной температуре. Если изотерма сорбции линейна, установление равновесия происходит мгновенно и степень диффузии вещества в подвижной фазе пренебрежимо мала, идеальный хроматографический пик описывается кривой нормального распределения.
Для объяснения причин размывания хроматографических зон используются две теории: теоретических тарелок и кинетическая теория.
Теория теоретических тарелок предполагает, что:
• каждая хроматографическая колонка состоит из некоторого количества одинаковых по величине абстрактных узких слоёв, называемых теоретическими тарелками, на каждой тарелке происходит один элементарный акт сорбции-десорбции;
• на каждой тарелке происходит мгновенное установление равновесия между веществом, находящимся в подвижной и неподвижной фазе;
• переход вещества с одной тарелки на другую происходит дискретно - при попадании на тарелку новой порции элюента равновесие нарушается, и часть вещества мгновенно переносится на следующую тарелку, где вновь мгновенно наступает равновесие и т. д.;
• на любой тарелке в любой момент времени число сорбируемых частиц вещества значительно больше числа сорбируемых частиц растворителя, изотерма сорбции является линейной.
Количественной характеристикой хроматографической колонки являются: высота эквивалентная теоретической тарелке (H) и число теоретических тарелок (N).
| н=L N |
| N=L H |
Высота эквивалентная теоретической тарелке представляет собой дисперсию, приходящуюся на единицу длины колонки. Чем
меньше H и больше N, тем в меньшей степени происходит размывание пика и тем эффективнее хроматографическое разделение (рис. 22.4).

|
Рис. 22.4. Хроматограммы вещества X, полученные на колонках с различной эффективностью (размерность оси абсцисс одинакова)
|
| где tR - время удерживания, kx - коэффициент, величина которого зависит от того, на каком уровне измеряется ширина пика wx. |
Если пик представляет собой кривую нормального распределения, то ширина пика у основания равна 4а, на половине высоты - 2,35а, на 60,7% высоты (между точками перегиба) - 2а (рис. 22.5) и
т.д. При измерении ширины пика у основания коэффициент kx бу-
2 2 дет равен 16 (4), на половине высоты - 5,54 (2,35) и т.д.

|
| h 0,607h 0,5h |
| Рис. 22.5. Свойства идеального хроматографического пика |
Число теоретических тарелок является мерой эффективности колонки и обычно постоянно для всех пиков на хроматограмме. Так как N является постоянной величиной, то при увеличении времени удерживания ширина пика увеличивается.
| Число теоретических тарелок можно рассчитать: |
288
Согласно кинетической теории хроматографии размывание хроматографических пиков обусловлено одновременным действием трёх независимых друг от друга процессов:

| продольная диффузия ] |
| B |
РАЗМЫВАНИЕ ХРОМАТОГРАФИЧЕСКОГО ПИКА
сопротивление массопереносу^ C

| В насадочной колонке молекулы компонентов разделяемой пробы движутся между частицами сорбента по разным траекториям. |
В процессе движения полосы вещества по колонке его молекулы непрерывно переносятся из подвижной фазы в неподвижную и обратно. Процесс переноса требует определённого времени.
Л
вихревая диффузия ] A
| влияние | |
| / / 1 * -——_ | не зависит |
| от скорости | |
| / / / / ч/ | ПФ |
Суммарное влияние вихревой диффузии, продольной диффузии и сопротивления массопереносу на величину высоты эквивалентной теоретической тарелке описывается уравнением Ван-Деемтера.
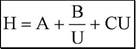
|
где U - линейная скорость подвижной фазы

|
| H |
| CU |
| B/U |
| опт U Рис. 22.6. Зависимость H от линейной скорости газа-носителя |
| H |
| U |
Зависимость H от U для газовой хроматографии (насадочная колонка) показана на рис. 22.6. Оптимальную скорость газа-носителя, при которой величина Н минимальна, можно рассчитать по формуле:
| B | |
| U опт ^ | C |
В жидкостной хроматографии величина B практически не вносит вклад в размывание хроматографического пика (вязкость жидкости значительно больше вязкости газа), поэтому зависимость Н от Uвыглядит по-другому (как?).
Для характеристики эффективности разделения компонентов смеси, используют коэффициент разделения (а) и разрешение (RS).

|
| Если a = 1, то разделение невозможно. Разрешение рассчитывается по следующей формуле: |

|
Коэффициент разделения равен отношению исправленных времён удерживания (а также VR, k', D) веществ:
Разделение двух пиков считается полным, если RS > 1,5 (при RS = 1,5 расстояние между максимумами пиков составляет 6а, степень перекрывания пиков 0,13%) - рис. 22.6.
Rs=_L6l=1,5 S 4а + 4а
 Рис. 22.6. Перекрывание пиков при различной величине RS
Рис. 22.6. Перекрывание пиков при различной величине RS
|
Число теоретических тарелок, необходимое для разделения с заданным разрешением, равно:
| N = 16R2 | f k 2 +1 1 | f a 1 | ||
| I k 2 J | la-1J |
ГЛАВА 23
ГАЗОВАЯ ХРОМАТОГРАФИЯ
23.1. Общая характеристика
Газовая хроматография - группа хроматографических методов, в которых подвижная фаза газообразна (находится в состоянии газа или пара).

|
| газ ожидкостная |
В зависимости от агрегатного состояния неподвижной фазы:
ГАЗОВАЯ ХРОМАТОГРАФИЯ

|
| ГЖХ |
газотвёрдофазная (газоадсорбционная)
неподвижной фазой является дисперсное твёрдое тело (адсорбент) неподвижной фазой является слой жидкости, нанесённой на поверхность твёрдого носителя (зернистый мелкодисперсный материал или внутренние стенки колонки)
23.2. Устройство газового хроматографа
Газохроматографические определения проводятся с помощью прибора, называемого газовым хроматографом. Принципиальная схема такого прибора приведена на рис. 23.1.
ввод пробы
 Рис. 23.1. Принципиальная схема газового хроматографа
Рис. 23.1. Принципиальная схема газового хроматографа
|
В ГАХ и ГЖХ используется один и тот же прибор. Различие между данными вариантами газовой хроматографии заключается лишь в содержимом хроматографической колонки.
Газожидкостного хроматографа не существует!
Подвижная фаза (газ-носитель)
В качестве подвижной фазы в газовой хроматографии применяют азот, гелий, водород, аргон и другие вещества. Газ-носитель должен:
• быть инертен по отношению к определяемым веществам и
сорбенту;
• иметь как можно меньшую вязкость;
• обеспечивать высокую чувствительность детектора;
• быть доступным, взрывобезопасным, достаточно чистым и
т.д.
Газы-носители хранятся в стальных баллонах под давлением (до 150 атм). Газ отбирается из баллона с помощью редуктора (устройства, позволяющего отбирать газ из баллона при давлении намного меньшем, чем давление в баллоне). Система подготовки газа необходима для установки, стабилизации, очистки газовых потоков, а также измерения их скорости. Она включает в себя регулятор давления, регулятор расхода газа, фильтры для очистки газа и т.д.
Дата добавления: 2015-09-07; просмотров: 296 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| Основные характеристики внешней хроматограммы, получаемой при элюентном хроматографическом анализе | | | Хроматографическая колонка |