Читайте также:
|
SUPPLEMENTARY TEXTS
Principles of Stereochemistry
For most combinations of atoms, a number of molecular structures that differ from each other in the sequence of bonding of the atoms are possible. Each individual molecular assembly is called an isomer, and the constitution of a compound is the particular combination of bonds between atoms (molecular connectivity) which is characteristic of that structure. Propanal,allyl alcohol, acetone, 2-methyloxirane, and cyclopropanol each correspond to the molecular formula C3H6O, but differ in constitution and are isomers of one another.
When structures having the same constitution differ with respect to their spatial arrangement, they are stereoisomers.Stereoisomers are described by specifying their topology and the nature of their relationship to other stereoisomers of the same constitution. Stereoisomers differ in configuration,and in order to distinguish between stereoisomeric compounds, it is necessary to specify the configuration. If two stereoisomers are nonsuperimposable mirror images, the molecules are enantiomers. Structures which have nonsuperimposable mirror images are called chiral. Chirality is the property of any molecule (or other object) of being nonsuperimposable on its mirror image. Samples which contain only one enantiomer are called enantiomerically pure or homochiral. Stereoisomers which are not enantiomers are diastereomers.
It is possible to obtain pure enantiomers of chiral compounds. One property of separated enantiomers is to cause the rotation of the plane of polarized fight by oppositebut equal amounts. Samples mat contain equal amounts of two enantiomers have zero net rotation and are called racemic mixtures. Samples that contain only one of the enantiomers are said to be enantiomerically pure. Samples that have an excess of one enantiomer over the other are enantiomerically enriched and show a net rotation of polarized light and are said to be optically active.
In addition to constitution and configuration, there is a third important level of structure, that of conformation. Conformations are discrete molecular arrangements that differ in spatial arrangement as a result of facile rotations about single bonds. Usually, conformers are in thermal equilibrium and cannot be separated. A special case of stereoisomerism arises when rotation about single bonds is sufficiently restricted by steric or other factors that-the different conformations can be separated. The term atropisomeris applied to stereoisomers that result from restricted bond rotation.
Enantiomeric Relationships
The relationship between chirality and optical activity is historically such a close one that chemists sometimes use the terms imprecisely. Optical activity refers to just one property of chiral molecules, namely, the ability to rotate plane-polarized light Measurement of optical activity is useful both for determining the configuration of chiral molecules and for investigating the stereochemical relationship between reactants and products. The mechanics of measuring optical rotation will not be discussed here since the basic method is described in most introductory texts. Both the sign and the magnitude of optical rotation are dependent on the conditions of the measurement, including temperature, solvent, and the wavelength of the light By convention, single-wavelength measurements are usually made at the 589-nm emission line of sodium arc lamps. This wavelength is known as the sodium D line, and optical rotations measured at this wavelength are designated [ɑ]D.
Pure enantiomeric substances show rotations that are equal in magnitude but opposite in direction. Unequal mixtures of enantiomers rotate light in proportion to the composition. The relationship between optical purity and measured rotation is
Optical purity (%) = 

The optical purity is numerically equivalent to the enantiomeric excess (e.e.), which is defined as

Measurement of rotation as a function of wavelength is useful in structural studies aimed at determining the chirality of a molecule. This technique is called optical rotatory dispersion (ORD). The resulting plot of rotation against wavelength is called an ORD curve. The shape of the ORD curve is determined by the configuration of the molecule and its absorption spectrum. In many cases, the ORD curve can be used to specify the configuration of a molecule by relating it to those of similar molecules of known configuration. Chiral substances also show differential absorption of circularly polarized light This is called circular dichroism(CD) and is quantitatively expressed as the molecular ellipticity, Ө:

where ԑL and ԑRare the extinction coefficients of left and right circularly polarized light.
The molecular ellipticity is analogous to specific rotation in that two enantiomers have exactly opposite values of Өat every wavelength. Two enantiomers will thus show CD spectra having opposite signs. A compound with several absorption bands may show both positive and negative bands.
Although measurements of optical rotation and ORD or CD spectra have historically been the main methods for determining enantiomeric purity and assisting configuration, other analytical techniques are also available. High-performance liquid chromatography (HPLC) using chiral column packing material can resolve enantiomers on both an analytical and a preparative scale. Chiral packing materials for gas-liquid chromatography (GLC) have also been developed.
Compounds in which one or more carbon atoms have four nonidentical substituents are the largest class of chiral molecules, Carbon atoms with four nonidentical ligands are referred to as asymmetric carbon atoms because the molecular environment at such a carbon atom possesses no element of symmetry. Asymmetric carbons are a specific example of a stereogenic center. A stereogenic center is any structural feature that gives rise to chirality in a molecule. 2-Butanol is an example of a chiral molecule and exists as two nonsuperimposable mirror images. Carbon-2 is a stereogenic center.

Ethanol is an achiral molecule. The plane defined by atoms C-1,C-2, and O is a plane of symmetry. Any carbon atom with two identical ligands contains a plane of symmetry that includes the two nonidentical ligands. Any molecule, no matter how complex, that possesses a plane of symmetry is achiral.

There are a number of important kinds of stereogenic centers besides asymmetric carbon atoms. One example is furnished by sulfoxides with nonidentical substituents on sulfur. Sulfoxides are pyramidal and maintain their configuration at room temperature. Unsymmetrical sulfoxides are therefore chiral and exist as enantiomers. Sulfonium salts with three nonidentical ligands are also chiral as a result of their pyramidal shape.
Although unsymmetrically substituted amines are chiral, the configuration is not stable because of rapid inversion at nitrogen. The activation energy for pyramidal inversion at phosphorus is much higher than at nitrogen, and many optically active phosphines have been prepared. The barrier to inversion is usually in the range of 30-35 kcal/mol so that enantiomerically pure phosphines are stable at room temperature but racemize by inversion at elevated temperatures. Asymmetrically substituted tetracoordinate phosphorus compounds such as phosphonium sails and phosphine oxides are also chiral.
The chirality of a molecule is described by specifying its configuration. The system that is used is the Cahn-Ingold-Prelogconvention, which uses the descriptors Rand S.The Fischer convention,employing the descriptors D and L, is historically important and is still used with certain types of molecules.
The Cahn Ingold-Prelog descriptors Rand Sare assigned by using the sequence rideto assign a priority order to the substituents on the atom to which a configuration is being assigned The substituent atoms are assigned decreasing priority in the order of decreasing atomic number. When two or more of the substituent atoms are the same dement (e.g., carbon), the assignment of priority is based on the next attached atom in those substituents. This process is continued until the order of priority of all substituents has been established. An atom that is multiply bonded is counted once for each formal bond. When the substituent priority has been established, the molecule is viewed in an orientation that places the lowest-priority substituent behind the stereogenic center. The three remaining substituents project toward the viewer. The remaining substituents have one of two possible arrangements. The substituents decrease in priority in either a clockwise manner or in a counterclockwise manner. In the former case, the configuration R(for rectus)is assigned. If the priority decreases in the counterclockwise sense, the atom is of S (for sinister)configuration.
The configuration of the 2-butanol enantiomer shown below is established as Sas follows. The highest-priority atom bonded to the asymmetric carbon is O; the lowest is H. The remaining two atoms are each C, and the choice as to which of these is of higher priority is made by comparing their ligands. The methyl group has (H, H, H), while the ethyl group has (С, H, H); therefore, the ethyl group is of higher priority than the methyl group. The complete priority list is; OH > C 2H 5> CH3> H. When viewed from the side opposite the lowest-priority ligand, the remaining groups appear in order of decreasing priority in counterclockwise fashion, and the configuration is S

Some other examples of assignment of configuration are illustrated below.

When a stereogenic center is tricoordinate, as is the case for sulfoxides, sulfonium salts, and phosphines, then a "phantom atom" of atomic number zero is taken to occupy the lowest-priority site of a presumed tetrahedral atom.
Glyceraldehye is the point of reference for describing the configuration of carbohydrates and other natural substances in accordance with the Fischer convention.The two enantiomers were originally arbitrarily assigned the configurations d and l as shown below. Subsequently, a determination of the configuration of sodium rubidium tartrate by X-ray crystallography and the relationship of this material to D-glyceraldehyde established that the original arbitrary assignments were the correct ones.

In the Fischer convention, the configuration is of other molecules are described by the descriptors d and l, which are assigned by comparison with the reference molecule glyceraldehyde. In employing the Fischer convention, it is convenient to use projection formulas.These are planar representations Defined in such a way as to convey three-dimensional structural information. The molecule is oriented with the major carbon chain aligned vertically in such a manner mat the most oxidized terminal carbon is at the top. The vertical bonds at each carbon are directed back, away from the viewer, and the horizontal bonds are directed toward the viewer. The D and L forms of glyceraldehyde are shown below with the equivalent Fischer projection formulas.

The assignment of the configuration of any other chiral molecule in the Fischer convention is done by comparison with D- and L-glyceraldehyde. The molecule is aligned with the chain vertical and the most oxidized carbon at the top, as specified by the Fischer convention. The stereogenic center with the highest number (at the lowest position in the Fischer projection) is compared with C-2of glyceraldehyde. If the configuration is that of D-glyceraldehye, the molecule is assigned the D-configuration, whereas if it is like that of L-glyceraldehyde, it is assigned the L-configuration. This is illustrated below with several carbohydrates.

The amino acids found in proteins have the L-configuration, as illustrated for alanine, serine, and leucine.

At the present time, use of the Fischer convention is almost entirely restricted to carbohydrates, amino acids, and biologically important molecules of closely related structural types. The problem with more general use is that there are no adequate rules for deciding whether a chiral atom is "tike" D-glyceraldehyde or L-glyceraldehyde when the structures are not closely similar to the reference molecules. This relationship is clear for carbohydrates and amino acids.
The property of chirality is determined by overall molecular topology, and there are many molecules that are chiral even though they do not possess an asymmetrically substituted atom. The chirality of E-cyclooctene and Z,E-cyclooctadiene is also dependent on restricted rotation. Manipulation of a molecular model will illustrate that each of these molecules can be converted into its enantiomer by a rotational process by which the ring is turned "inside-out."
There is no direct relationship between the configurational descriptors R and S or D and l and the sign of rotation of the molecule. Ror Smolecules can have either + or - signs for rotation, as can d or L molecules. Thus, even though a configuration can be specified on the basis of these conventions, additional information is necessary to establish which molecule of an enantiomeric pair possesses the specified configuration. Determination of the absolute configurationestablishes the configuration of each enantiomer. There are several approaches to this problem. One is to establish a direct structural relationship to a molecule of known configuration by chemical transformation. This is the way in which most of the reference molecules whose absolute configurations are known were initially assigned. The existence of a base of molecules whose absolute configurations are known has permitted the development of correlations based on the CD and ORD curves of certain types of chromophores. When chromophores are located close to stereogenic centers, the spectroscopic properties are affected in a predictable way so that the sign and shape of the ORD or CD curve can be a reliable basis for configurational assignment. While routine X-ray crystal structure determination does not provide the absolute configuration of the molecule, special analysis of the diffraction data does allow assignment of absolute configuration. These methods are important, for example, in assigning the absolute configuration of new natural products.

Diastereomeric Relationships
Diastereomers include all stereoisomers that are not related as an object and its mirror image. These structures represent the four stereoisomers of 2,3,4-trihydroxybutanal. The configurations of C-2 and C-3 are indicated. Each stereogenic center is designated Ror S by application of the sequence rule. Each of the four structures is stereoisomeric with respect to any of the others. The 2R,3Rand 2S,3Sisomers are enantiomeric, as are the 2R,3Sand 2S,3Rpair. The 2R,3S isomer is diastereomeric with the 2S,3S and 2R,3Risomers because they are stereoisomers but not enantiomers. Any given structure can have only one enantiomer. All other stereoisomers of that molecule are diastereomeric. The relative configuration of diastereomeric molecules is frequently specified using the terms synand anti.The molecules are represented as extended chains. Diastereomers with substituents on the same side of the extended chain are synstereoisomers, whereas those with substituents on opposite sides are anti stereoisomers.
Diastereoisomers differ in both physical properties and chemical reactivity. They generally have different melting points, boiling points, solubility, chromatographic mobility, and so on. The specific rotations of diastereomeric molecules differ in both magnitude and sign. The difference in chemical reactivity can be small, such as a difference in rate, or two diastereomers can lead to entirely different products, depending on the mechanism of the particular reaction. Because of their differing physical and chemical properties, diastereomers can be separated by methods such as crystallization or cinematography.
Sometimes the terms erythroand threoare used to specify the relative configuration of two adjacent stereogenic centers. The terms are derived torn the sugars erythrose and threose. The terms were originally defined such that a Fischer projection formula in which two adjacent substituents were on the same side was the erythroisomer and that in which the substituents were on opposite sides was the threoisomer.

Unfortunately, assignment of molecules that are not closely related to the reference molecules becomes a subjective matter of assigning which substituents are "similar". The application of the terminology to cases in which the chiral centers are not adjacent is also ambiguous. As a result, the threo—erythroterminology is not a general method of specifying stereochemical relationships.
Fischer projection formulas can be used to represent molecules with several stereogenic centers and are commonly used for carbohydrates. For other types of structures, a more common practice is to draw the molecule in an extended conformation with the main chain horizontal In this arrangement, each tetrahedral carbon has two additional substituents, one facing out and one in. The orientation is specified with solid wedged bonds for substituents facing out and with dashed bonds for substituents that point in.
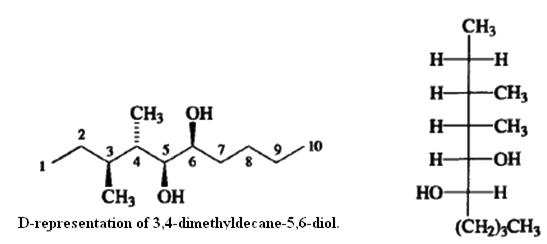
Since the main chain in this representation is in an entirely staggered conformation, whereas in the Fischer projection formulas the conformation represented is completely eclipsed, an antirelationship between two adjacent substituents in an extended structure corresponds to being on the same side in a Fischer projection formula (erythro)whereas a syn relationship corresponds to being on opposite sides in the Fischer projection (threo).
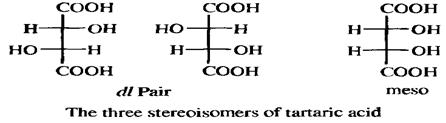
Since chirality is a property of a molecule as a whole, the specific juxtaposition of two or more stereogenic centers in a molecule may result in an achiral molecule. For example, there are three stereoisomers of tartaric acid (2,3-dihydroxybutanedioic acid), Two of these are chiral and optically active but the third is not.The reason that the third stereoisomer is achiral is that the substituents on the two asymmetric carbons are located with respect to each other in such a way that a molecular plane of symmetry exists.Compounds that incorporate asymmetric atoms but are nevertheless achiral are called mesaforms.

This situation occurs whenever pairs of stereogenic centers are disposed in the molecule in such a way as to create a plane of symmetry. A particularly striking example is the antibiotic nonactin.

Incorporation of stereogenic centers into cyclic structures produces special stereochemical circumstances. Except in the case of cyclopropane, the lowest-energy conformation of the rings is not planar. Most cyclohexane derivatives adopt a chair conformation. For example, the two conformers of cis-1,2-dimethylcyclohexane are both chiral. However, the two conformers are enantiomeric so the conformational change leads to racemization. Because the barrier to this conformational change is low (10kcal/mol) the two enantiomers are rapidly interconverted.

Certain dimethylcycloalkanes contain a plane of symmetry. For example, both chair conformers of cis-1,3-dimethylcyclohexane possess a plane of symmetry bisecting the molecule through C-2 and C-5. The transisomer does not have any element of symmetry and is chiral.

One simple test for chirality of substituted cycloalkanes is to represent the ring in planar form. If the planar form is achiral because of a symmetry element, the compound will not exist as an enantiomerically biased sample, even if individual conformers may be chiral.
Since the presence of a plane of symmetry in a molecule ensures that it will be achiral, one approach to classification of stereoisomers as chiral or achiral is to examine the molecule for symmetry elements. There are other elements of symmetry in addition to planes of symmetry that ensure that a molecule will be superimposable on its mirror image. The trans, cis, cisand trans, trans, cisstereoisomers of 1,3-dibromo-trans-2,4-dimethylcyclobutane are illustrative, This molecule does not possess a plane of symmetry, but the mirror images are superimposable, as illustrated below. This molecule possesses a center ofsymmetry.A center of symmetry is a point from which any line drawn through the molecule encounters an identical environment in either direction from the center of symmetry.


Because diastereoisomers have different physical and chemical properties, they can be separated by a range of chemical and physical methods. The process of resolutionis the separation of a racemic mixture. Separation is frequently effected by converting the enantiomers into a mixture of diastereomers by reaction with a pure enantiomer of a second reagent, the resolving agent.Because the two resulting products will be diastereomeric, they can be separated. The separated diastereomers can then be reconverted to the pure enantiomers by reversing the initial chemical transformation. An example of this method is shown in Scheme for the resolution of a racemic carboxylic acid by way of a diastereomeric salt resulting from reaction with an enantiomerically pure amine. The R-acid, R-amine and S-acid, S-amine salts are separated by fractional recrystallization. The resolved acids are regenerated by reaction with a strong acid, which liberates the carboxylic acid from the amine salt.

Although the traditional method of separating the diastereomeric compounds generated in a resolution procedure is fractional crystallization, chromatographic procedures are now common and convenient. Diastereomeric compounds exhibit different adsorption on achiral materials and can be separated by column chromatography or by taking advantage of the greater separation powers of HPLC.
Separation of enantiomers by physical or chemical methods requires the use of a chiral material, reagent, or catalyst. Both natural materials, such as polysaccharides and proteins, and solids that have been synthetically modified to incorporate chiral structures have been developed for use in separation of enantiomers by HPLC. The use of a chiral stationary phase makes the interactions between the two enantiomers with the adsorbent nonidentical and thus establishes a different rate of elution through the column. The interactions typically include hydrogen bonding, dipolar interactions, and π-π interactions. These attractive interactions may be disturbed by steric repulsions, and frequently the basis of enantioselectivity is a better steric fit for one of the two enantiomers.
The potential for use of chiral natural materials such as cellulose for separation of enantiomers has long been recognized, but development of efficient materials occurred relatively recently. Several acylated derivatives of cellulose are effective chiral stationary phases. Benzoate esters and aryl carbamates are particularly useful. These materials are commercially available on a silica support and under the trademark Chiralcel.
Synthetic chiral adsorbents are usually prepared by tethering a chiral molecule to a silica surface. The attachment to the silica is through alkylsiloxy bonds. A study which demonstrates the technique reports the resolution of a number of aromatic compounds on a 1- to 8-g scale. The adsorbent is a silica that has been derivatized with a chiral reagent. Specifically, hydroxyl groups on the silica surface are covalently bound to a derivative of R-phenylglycine.

A medium-pressure chromatography apparatus is used. The racemic mixture is passed through the column, and, when resolution is successful, the separated enantiomers are isolated as completely resolved fractions.
Another means of resolution depends on the difference in rates of reaction of two enantiomers with a chiral reagent. The transition-state energies for reaction of each enantiomer with one enantiomer of a chiral reagent will be different. This is because the transition states and intermediates (R-substrate… R-reactant) and (S-substrate…S-reactant) are diastereomeric. Kinetic resolutionis the term used to describe the separation of enantiomers based on different reaction rates with an enantiomerically pure reagent.

Figure 2.5 summarizes the basis of kinetic resolution. Because the separation is based on differing rates of reaction, the degree of resolution that can be achieved depends on both the magnitude of the rate difference and the extent of reaction. The greater the difference in the two rates, the higher the enantiomeric purity of both the reacted and the unreacted enantiomer. The extent of enantiomeric purity can be controlled by controlling the degree of conversion. As the degree of conversion increases, the enantiomeric purity of the unreacted enantiomer becomes very high.


The relationship between the relative rate of reaction, extent of conversion, and enantiomeric purity of the unreacted enantiomer is shown in Fig. 2.6. Of course, the high conversion required for high enantiomeric purity when the relative reactivity difference is low has a serious drawback. The yieldof the unreactedsubstrate is low if the overall conversion is high Thus, with relative reactivity differences of < 10, high enantiomeric purity can be achieved only at the expense of low yield.

Preparation of enantiomerically enriched materials by use of chiral catalysts is also based on differences in transition-state energies. While the reactant is part of a complex or intermediate containing a chiral catalyst, it is in a chiral environment The intermediates and complexes containing each enantiomeric reactant and a homochiral catalyst are diastereomeric and differ in energy, This energy difference can then control selection between the stereoisomeric products of the reaction. If the reaction creates a new stereogenic center in the reactant molecule, there can be a preference for formation of one enantiomer over the other.
Enzymes constitute a particularly important group of enantioselective catalysts. Enzymes are highly efficient and selective catalysts and can carry out a variety of transformations. Because the enzymes are derived from L-amino acids, they are homochiral, and usually one enantiomer of a reactant is much more reactive than the other. The reason is that the interaction of the enzyme with one enantiomer is diastereomeric to its interaction with the other. Because enzyme catalysis is usually based on a specific fit to an "active site," the degree of selection between the two enantiomers is often very high. Enzyme-catalyzed reactions can therefore be used to resolve organic compounds. The most completely characterized enzymes that are available are those which catalyze hydrolysis of esters and amides (esterases, lipases, peptidases, acylases) and those which oxidize alcohols to ketones or aldehydes (dehydrogenases). Purified enzymes can be used, or the reaction can be done by incubating the reactant with an organism (yeast, for example) that produces an appropriate enzyme during fermentation.
The differing physical properties of diastereomers are also the basis for a particularly sensitive method for assessing the enantiomeric purity of compounds. Although, in principle, enantiomeric purity can be determined by measuring the optical rotation, this method is reliable only if the rotation of the pure compound is accurately known. This is never the case for a newly prepared material and is often uncertain for previously prepared compounds. If a derivative of a chiral compound is prepared in which a new chiral center is introduced, the two enantiomers will give different diastereomers. Because these will have different physical properties, their relative amounts can be determined. NMR spectroscopy is a convenient means of detecting and quantitating the two diastereomeric products. A pure enantiomer will give only a single spectrum, but a partially resolved material will show two overlapping spectra in the ratio of the two diastereomeric derivatives. The most widely used derivatizing reagent for the NMR method is a compound known as Mother's reagent.One reason that this compound is particularly useful is mat the aromatic ring usually induces markedly different chemical shifts in the two diastereomeric products that are formed.

Changes in NMR spectra can also be observed as the result of formation of noncovalent complexes between enantiomeric molecules and another chiral reagent. This is the basis of the use of chiral shift reagentsto determine the enantiomeric purity of chiral substances. Several of the lanthanide elements have the property of forming strong complexes with alcohols, ketones, and other functional groups having Lewis base character. Ifthe lanthanide ion is in a chiral environment as the result of an enantiomerically pure ligand, two diastereomeric complexes are formed. The lanthanide elements induce large NMR shifts, and, as a result, shifted spectra are seen for the two complexed enantiomers. The relative intensities of the two spectra correspond to the ratio of enantiomers present in the sample.



Geometric isomers of alkenes are diastereomeric, since they are stereoisomers but not enantiomeric. The specification of the geometry of double bonds as cisand transsuffers from the same ambiguity as specifying configuration by the Fischer convention; that is, it requires a subjective judgment about the "similarity'" of groups. The sequence rule is the basis for an unambiguous method for assignment of alkene geometry. The four substituents on the double bond are taken in pairs. The sequence rules are used to determine if the higher-priority groups on the atoms forming the double bond are on the same side or opposite sides of the double bond.

If the higher-priority groups are on the same side, the descriptor is Z(from the German word zusammen,together); if they are on opposite sides, the descriptor is E(from entgegen,opposite). As in applying the sequence rule to stereogenic centers, if the atoms directly attached to the double bond have the same atomic number, the priorities are assigned by sequentially comparing atoms in the substituent until priority can be established. The system can also be applied to multiple bonds involving elements other than carbon, such as C=N. The Zand Edescriptors have replaced synand antifor describing the stereochemistry of oximes. As in the case of stereogenic centers, if an atom at a double bond does not have two substituents (as is the case for oximes), then a "phantom ligand" with atomic, number zero is assumed and assigned the lower priority. Scheme 2.8 shows some stereoisomers compounds named according to the sequence rule convention.

METHODS OF DETERMINING CONFIGURATION
In all the methods, it is necessary to relate the compound of unknown configuration to another whose configuration is known. The most important methods of doing this are:
1. Conversion of the unknown to, or formation of the unknown from, a compound of known configuration without disturbing the chiral center. Since the chiral

center was not disturbed, the unknown obviously has the same configuration as the known. This does not necessarily mean that if the known is (R), the unknown is also (R). For example, when (R)-l-bromo-2-butanol is reduced to 2-butanol without disturbing the chiral center, the product is the (S) isomer, even though the configuration is unchanged, because CH3CH2 ranks lower than BrCH2 but higher than CH3.
2. Conversion at the chiral center if the mechanism is known. Thus, the Sn2 mechanism proceeds with inversion of configuration at an asymmetric carbon. It was by a series of such transformations that lactic acid was related to alanine:
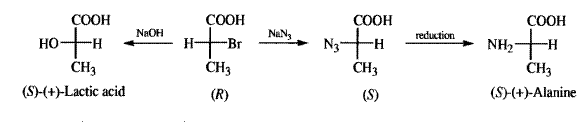
3. Biochemical methods. In a series of similar compounds, such as amino acids or certain types of steroids, a given enzyme will usually attack only molecules with one kind of configuration. If the enzyme attacks only the l form of eight amino acids, say, then attack on the unknown ninth amino acid will also be on the l form.
4. Optical comparison. It is sometimes possible to use the sign and extent of rotation to determine which isomer has which configuration. In a homologous series, the rotation usually changes gradually and in one direction. If the configurations of enough members of the series are known, the configurations of the missing ones can be determined by extrapolation. Also, certain groups contribute more or less fixed amounts to the rotation of the parent molecule, especially when the parent is a rigid system such as a steroid.
5. The special X-ray method of Bijvoet gives direct answers and has been used in a number of cases.
6. One of the most useful methods for determining enantiomeric composition is to derivatize the alcohol with a chiral nonracemic reagent and examine the ratio of resulting diastereomers by gas chromatography. There are many derivatizing agents available, but the most widely used are derivatives of ɑ-methoxy-ɑ -trifluoromethylphenyl acetic acid (MTPA, or Mosher's acid, 38).Reaction with a chiral nonracemic alcohol (R*OH) generates a Mosher's ester (39) that can be analyzed for diastereomeric composition by 1H or 19F NMR as well as by chromatographic techniques.

Alternatively, complexation with lanthanide shift reagents allow the signals of the MTPA ester to be resolved and used to determine enantiomeric composition. The effectiveness of this method, and other related methods, have been evaluated and found to be good for determining the absolute configuration of the alcohol of interest (R*OH). Two, of many other reagents that have been developed to allow the enantiopurity of alcohols and amines to be determined include 40 and 41. Chloromethyl lactam (40) reacts with R*OH or R*NHR (R*NH2), forming derivatives that allow analysis by 1H NMR and 41 reacts with alkoxides (R*O-)100 to form a derivative that can be analyzed by 31P NMR.

Other methods have also been used, including optical rotatory dispersion, circular dichroism (CD), and asymmetric synthesis.
The Cause of Optical Activity
The question may be asked: Just why does a chiral molecule rotate the plane of polarized light? Theoretically, the answer to this question is known and in a greatly simplified form may be explained as follows.
Whenever any light hits any molecule in a transparent material, the light is slowed because of interaction with the molecule. This phenomenon on a gross scale is responsible for the refraction of light and the decrease in velocity is proportional to the refractive index of the material. The extent of interaction depends on the polarizability of the molecule. Plane-polarized light may be regarded as being made up of two kinds of circularly polarized light. Circularly polarized light has the appearance (or would have, if one could see the wave) of a helix propagating around the axis of light motion, and one kind is a left-handed and the other a right-handed helix. As long as the plane-polarized light is passing through a symmetrical region, the two circularly polarized components travel at the same speed. However, a chiral molecule has a different polarizability depending on whether it is approached from the left or the right. One circularly polarized component approaches the molecule, so to speak, from the left and sees a different polarizability (hence, on a gross scale, a different refractive index) than the other and is slowed to a different extent. This would seem to mean that the left- and right-handed circularly polarized components travel at different velocities, since each has been slowed to a different extent. However, it is not possible for two components of the same light to be traveling at different velocities. What actually takes place, therefore, is that the faster component "pulls" the other toward it, resulting in rotation of the plane. Empirical methods for the prediction of the sign and amount of rotation based on bond refractions and polarizabilities of groups in a molecule have been devised, and have given fairly good results in many cases.
In liquids and gases, the molecules are randomly oriented. A molecule that is optically inactive because it has a plane of symmetry will very seldom be oriented so that the plane of the polarized light coincides with the plane of symmetry. When it is so oriented, that particular molecule does not rotate the plane but all others not oriented in that manner do rotate the plane, even though the molecules are achiral. There is no net rotation because, the molecules being present in large number and randomly oriented, there will always be another molecule later on in the path of the light that is oriented exactly opposite and will rotate the plane back again. Even though nearly all molecules rotate the plane individually, the total rotation is zero. For chiral molecules, however (if there is no racemic mixture), no opposite orientation is present and there is a net rotation.
An interesting phenomenon was observed when the CD of chiral molecules was measured in achiral solvents. The chiral solvent contributed as much as 10-20% to the CD intensity in some cases. Apparently, the chiral compound can induce a solvation structure that is chiral, even when the solvent molecules themselves are achiral.
Molecules With More Than One
Chiral (Stereogenic) Center
When a molecule has two stereogenic centers, each has its own configuration and can be classified (R)or (S) by the Cahn-Ingold-Prelog method. There are a total of four isomers, since the first center may be (R)or (S)and so may the second. Since a molecule can have only one mirror image, only one of the other three can be the enantiomer of A. This is B [the mirror image of an (R)center is always an (S) center]. The compounds C and D are a second pair of enantiomers and the

relationship of C and D to A and B is designated by the term diastereomer. Diastereomers may be defined as stereoisomers that are not enantiomers. Being enantiomers, C and D must have identical properties; the same is true for A and B. However, the properties of A and B are not identical with those of C and D. They have different melting points, boiling points, solubilities, reactivity, and all other physical, chemical, and spectral properties. The properties are usually similar but not identical. In particular, diastereomers have different specific rotations; indeed one diastereomer may be chiral and rotate the plane of polarized light while another may be achiral and not rotate at all (an example is presented below).
It is now possible to see why enantiomers react at different rates with other chiral molecules but at the same rate with achiral molecules. In the latter case, the activated complex formed from the (R)enantiomer and the other molecule is the mirror image of the activated complex formed from the (S)enantiomer and the other molecule. Since the two activated complexes are enantiomeric, their energies are the same and the rates of the reactions in which they are formed must be the same. However, when an (R)enantiomer reacts with a chiral molecule that has, say, the (R)configuration, the activated complex has two chiral centers with configurations (R)and (R),while the activated complex formed from the (S)enantiomer has the configurations (S)and (R). The two activated complexes are diastereomeric, do not have the same energies, and consequently are formed at different rates.
Although four is the maximum possible number of isomers when the compound has two chiral centers (chiral compounds without a chiral carbon, or with one chiral carbon and another type of chiral center, also follow the rules described here), some compounds have fewer. When the three groups on one chiral atom are the same as those on the other, one of the isomers (called a meso form) has a plane of symmetry, and hence is optically inactive, even though it has two chiral carbons. Tartaric acid is a typical case. There are only three isomers of tartaric acid: a pair of enantiomers and an inactive meso form. For compounds that have two chiral atoms, meso forms are found only where the four groups on one of the chiral atoms are the same as those on the other chiral atom.

In most cases with more than two chiral centers, the number of isomers can be calculated from the formula 2n where n is the number of chiral centers, although in some cases the actual number is less than this, owing to meso forms. An interesting case is that of 2,3,4-pentanetriol (or any similar molecule). The middle carbon is not asymmetric when the 2- and 4-carbon atoms are both (R)[or both (S)] but is asymmetric when one of them is (R)and the other (S).Such a carbon is called a pseudoasymmetric carbon. In these cases, there are four isomers: two meso forms and one dl pair. The student should satisfy himself or herself, remembering the rules governing the use of the Fischer projections, that these isomers are different, that the meso forms are superimposable on their mirror images, and that there are no other stereoisomers. Two diastereomers that have a different configuration at only one chiral center are called epimers.
In compounds with two or more chiral centers, the absolute configuration must be separately determined for each center. The usual procedure is to determine the configuration at one center, and then to relate the configuration at that center to the others in the molecule. One method is X-ray crystallography, which cannot be used to determine the absolute configuration at any chiral center but which does give relative configurations of all the chiral centers in a molecule, and hence the absolute configurations of all once the first is independently determined. Other physical and chemical methods have also been used for this purpose.
The problem of how to name the different stereoisomers of a compound when there are more than two now arises. Enantiomers are virtually always called by the same name, being distinguished by (R)and (S) or d and l or (+) and (-). In the early days of organic chemistry, it was customary to give each pair of enantiomers a different name or at least a different prefix (such as epi-, peri-, etc.). Thus the aldohexoses are called glucose, mannose, idose, and so on, although they are all 2,3,4,5,6-pentahydroxyhexanal (in their open-chain forms). This practice was partially due to lack of knowledge about which isomers had which configurations. Today it is customary to describe each stereogenic position separately as either (R) or (S) or, in special fields, to use other symbols. Thus, in the case of steroids, groups above the "plane" of the ring system are designated β, and those below it ɑ. Solid lines are often used to depict βgroups and dashed lines for ɑ groups. An example is

For many open-chain compounds, prefixes are used that are derived from the names of the corresponding sugars and that describe the whole system rather than each chiral center separately. Two such common prefixes are erythro- and threo-, which are applied to systems containing two stereogenic carbons when two of the groups are the same and the third is different. The erythro pair has the identical


|
groups on the same side when drawn in the Fischer convention, and if Y were changed to Z, it would be meso. The threo pair has them on opposite sides, and if Y were changed to Z, it would still be a dl pair. Another system for designating stereoisomers uses the terms syn and anti. The "main chain" of the molecule is drawn in the common zigzag manner. Then, if two non-hydrogen substituents are on the same side of the plane defined by the main chain, the designation is syn; otherwise anti.
Asymmetric Synthesis
Organic chemists often wish to synthesize a chiral compound in the form of a single enantiomer or diastereomer, rather than as a mixture of stereoisomers. There are two basic ways in which this can be done. The first way, which is more common, is to begin with a single stereoisomer, and to use a synthesis that does not affect the chiral center (or centers), as in the glyceraldehyde-glyceric acid. The optically active starting compound can be obtained by a previous synthesis, or by resolution of a racemic mixture, but it is often more convenient to obtain it from nature, since many compounds, such as amino acids, sugars, and steroids are present in nature in the form of a single enantiomer or diastereomer. These compounds are regarded as a chiral pool; that is, readily available compounds that can be used as starting materials.
The other basic method is called asymmetric synthesis or stereoselective synthesis. As was mentioned before, optically active materials cannot be created from inactive starting materials and conditions; hence, true asymmetric synthesis is impossible, except in the manner previously noted. However, when a new stereogenic center is created, the two possible configurations need not be formed in equal amounts if anything is present that is not symmetric. We discuss asymmetric synthesis under four headings:
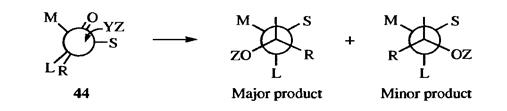
1. Active Substrate. If a new stereogenic center is created in a molecule that is already optically active, the two diastereomers are not (except fortuitously) formed in equal amounts. The reason is that the direction of attack by the reagent is determined by the groups already there. For certain additions to the carbon-oxygen double bond of ketones containing an asymmetric ɑ carbon, Cram's rule predicts which diastereomer will predominate (diastereoselectivity).

If the molecule is observed along its axis, it may be represented as in 44, where S, M, and L stand for small, medium, and large, respectively. The oxygen of the carbonyl orients itself between the small- and the medium-sized groups. The rule is that the incoming group preferentially attacks on the side of the plane containing the small group. By this rule, it can be predicted that 43 will be formed in larger amounts than 42.
Another model can be used to predict diastereoselectivity, which assumes reactant-like transition states and that the separation of the incoming group and any electronegative substituent at the a carbon is greatest. Transition state models 45 and 46 are used to predict diastereoselectivity in what is known as the Felkin-Ahn model.

Many reactions of this type are known, in some of which the extent of favoritism approaches 100%. The farther away the reaction site is from the stereogenic center, the less influence the latter has and the more equal the amounts of diastereomers formed.
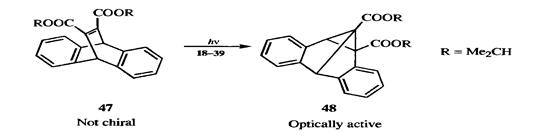
In a special case of this type of asymmetric synthesis, a compound (47) with achiral molecules, but whose crystals are chiral, was converted by UV light to a single enantiomer of a chiral product (48).
It is often possible to convert an achiral compound to a chiral compound by (1) addition of a chiral group; (2) running an asymmetric synthesis, and (3) cleavage of the original chiral group. An example is conversion of the achiral 2-pentanone to the chiral 4-methyl-3-heptanone (50). In this case, >99% of the product was the (S)enantiomer. Compound 49 is called a chiral auxiliary because it is used to induce asymmetry and then is removed.

Active Reagent. A pair of enantiomers can be separated by an active reagent that reacts faster with one of them than it does with the other (this is also a method of resolution). If the absolute configuration of the reagent is known, the configuration of the enantiomers can often be determined by a knowledge of the mechanism and by seeing which diastereomer is preferentiallyformed.

Creation of a new chiral center in an inactive molecule can also be accomplished with an active reagent, though it is rare for 100% selectivity to be observed. An example is the reduction of methyl benzoylformate with optically active N-benzyl-3-(hydroxymethyl)-4-methyl-1,4-dihydropyridine (51) to produce mandelic acid, which contained ~97.5% of the (S)-(+) isomer and 2.5% of the (R)-(-) isomer. Note that the other product, (52), is not chiral. Reactions like this, in which one reagent (in this case 51) gives up its chirality to another, are called selfimmolative. In this intramolecular example, chirality is transferred from one atom to another in the same molecule.

A reaction in which an inactive substrate is converted selectively to one of two enantiomers is called an enantioselective reaction, and the process is called asymmetric induction. These terms apply to reactions in this category and in categories 3 and 4.
When an optically active substrate reacts with an optically active reagent to form two new chiral centers, it is possible for both centers to be created in the desired sense. This type of process is called double asymmetric synthesis.
3. Active Catalyst or Solvent Many such examples are present in the literature, among them reduction of ketones and substituted alkenes to optically active (though not optically pure) secondary alcohols and substituted alkanes by treatment with hydrogen and a chiral homogeneous hydrogenation catalyst, the treatment of aldehydes or ketones with organometallic compounds in the presence of a chiral catalyst,and the conversion of alkenes to optically active epoxides by treatment with a hydroperoxide and a chiral catalyst. In some instances, notably in the homogeneous catalytic hydrogenation of alkenes, the ratio of enantiomers prepared in this way is as high as 98:2. Other examples of the use of a chiral catalyst or solvent are the conversion of chlorofumaric acid (in the form of its di-ion) to the (-)-threoisomer of the di-ion of chloromalic acid by treatment with H20 and the enzyme fumarase, and the preparation of optically active aldolsby the condensation of enolate anions with optically active substrates.
4. Reactions in the Presence of Circularly Polarized Light. If the light used to initiate a photochemical reaction of achiral reagents is circularly polarized, then, in theory, a chiral product richer in one enantiomer might be obtained. However, such experiments have not proved fruitful. In certain instances, the use of left- and right-circularly polarized light has given products with opposite rotations (showing that the principle is valid), but up to now the extent of favoritism has always been <1%.
Дата добавления: 2015-10-28; просмотров: 358 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| Mark and talk about five things from the text you are glad to find out about. Talk in pairs about these things and why you chose them. | | | HOW TO WRITE A REQUEST LETTER |