Читайте также:
|
В предыдущих параграфах мы рассмотрели несколько примеров, показывающих, что при определенных условиях каждая химическая реакция самопроизвольно протекает в определенном направлении. Так, при низких температурах экзотермическая реакция образования парообразной воды
2Н2 + О2 = 2Н2О+ 483,6 кДж
практически нацело протекает в прямом направлении. В отсутствие катализатора скорость этой реакции при обычных условиях крайне мала. Однако, при наличии катализатора (например, платинированного асбеста) процесс образования воды протекает с большой скоростью.
Но при высоких температурах эта реакция начинает идти в обратном направлении: водяной пар разлагается на водород и кислород. Во всех случаях в результате реакции может быть достигнуто состояние устойчивого химического равновесия, но само положение равновесия при разных условиях оказывается различным.
Возникает вопрос: в чем причина определенной направленности химических процессов, какие факторы обусловливают то или иное состояние химического равновесия?
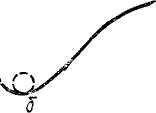
Известно, что в механических системах устойчивое равновесие соответствует минимуму потенциальной энергии системы. Так, шарик самопроизвольно скатывается из положения а на наклонной поверхности (рис. 69), причем его потенциальная энергия переходит сначала в кинетическую энергию движения шарика как целого, а затем в энергию теплового движения молекул. В положении б шарик находится в равновесии.
Естественно предположить, что и химические процессы должны самопроизвольно протекать в направлении уменьшения внутренней энергии системы, т. е. в направлении, отвечающем положительному тепловому эффекту реакции. Действительно, опыт показывает, что при обычных условиях самопроизвольно протекают преимущественно экзотермические реакции.
Однако попытка объяснить направленность химических процессов только стремлением к минимуму внутренней энергии приводит к противоречиям с фактами. Так, уже при обычных температурах самопроизвольно протекают эндотермические процессы растворения многих солей и некоторые эндотермические химические реакции.
Рис. 69. Шарик самопроизвольно скатывается из положения а в положение б.
|
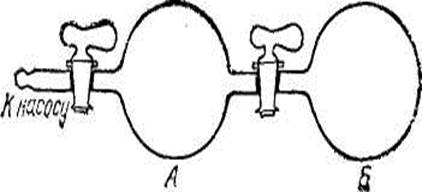
Рис. 70. Сосуд, состоящий из дзух частей: в части А находится разреженный газ, в части Б — вакуум.
|
С повышением температуры все большее число реакций начинает самопроизвольно протекать в направлении эндотермического процесса; примерами таких реакций могут служить упомянутое выше разложение воды или протекающий при высоких температурах синтез оксида азота(II):
0,5 N2 + 0,5 O2 = NO - 90,4 кДж
Более того, принцип стремления к минимуму внутренней энергии требует, чтобы все экзотермические реакции доходили до конца, т. е. исключает возможность обратимых реакций; однако такие реакции реально существуют.
Вспомним теперь, что среди механических систем имеются такие, поведение которых тоже нельзя описать только направленностью процессов к достижению минимума потенциальной энергии. Это системы, состоящие из очень большого числа частиц. Например, молекулы, входящие в состав воздуха, распределяются вокруг Земли в виде атмосферы многокилометровой толщины, но не падают на Землю, хотя минимуму потенциальной энергии каждой молекулы соответствует наиболее низкое ее положение.
Из громадного числа частиц состоят и химические системы. Поэтому неудивительно, что и здесь тенденция к достижению минимума внутренней энергии не является единственным фактором, определяющим их поведение.
Для того чтобы составить представление о втором факторе, влияющем на направление реакций, рассмотрим какой-либо самопроизвольно протекающий процесс, не сопровождающийся тепловым эффектом. Примером такого процесса может служить расширение разреженного газа.
Пусть в части А сосуда, разделенного на две части (рис. 70), находится разреженный газ. В таком газе среднее расстояние между молекулами велико; при этом условии внутренняя энергия газа не зависит от степени его разрежения. Вторая половина сосуда (Б) газа не содержит. Если открыть кран, соединяющий обе части сосуда, то газ самопроизвольно распространится по всему сосуду. Внутренняя энергия газа при этом не изменится; тем не менее, самопроизвольно произойдет именно процесс расширения газа, а обратный процесс — самопроизвольное сжатие газа — не происходит.
Причины такой направленности процесса можно понять, если сначала рассмотреть систему, содержащую небольшое число молекул. Пусть в сосуде находятся всего две молекулы, которые обозначим 1 и 2. Равномерное распределение газа между обеими частями сосуда, соответствующее определенному макросостоянию газа (Макросостояние — состояние вещества, характеризующееся определенными значениями его макроскопических свойств (температура, давление, объем и т. д.); микросостояние — состояние вещества, характеризующееся определенным состоянием каждой частицы (молекулы, атомы). Одно и то же макросостояние соответствует большому числу различных микросостояний) может осуществиться двумя м икросостояниями:
| А | Б |
Макросостояние, при котором весь газ сосредоточен в одной из частей сосуда (например, в части А), осуществляется единственным микросостоянием:
| А | Б |
| 1,2 | - |
Очевидно, что то или иное макросостояние системы тем более вероятно,
чем большим числом микросостояний оно может осуществиться.
В рассматриваемом случае (две молекулы) равномерное распределение
газа по сосуду вдвое вероятнее, чем переход всего газа в часть сосуда А.
Пусть теперь в сосуде находится четыре молекулы, которые мы вновь
пронумеруем. Переходу всего газа в часть сосуда А по-прежнему
соответствует единственное микросостояние:
| А | Б |
| 1,2,3,4 | - |
Между тем, равномерное распределение газа между обеими частями сосуда может теперь осуществляться шестью различными микросостояниями:
| А | Б | А | Б | А | Б |
| 1,2 1,3 | 3,4 2,4 | 1,4 2,3 | 2,3 1,4 | 2,4 3,4 | 1,3 1,2 |
Теперь, следовательно, вероятность равномерного распределения молекул газа по всему объему сосуда оказывается заметно более высокой, чем вероятность их перехода в одну из его частей. Естественно, что равномерное распределение газа будет наблюдаться гораздо чаще, чем полное его сосредоточение в части сосуда.
Если в сосуде находится шесть молекул, то переходу нх в одну из частей сосуда (т. е. самопроизвольному сжатию газа до половины занимаемого им первоначального объема), как и раньше, соответствует только одно микросостояние. Но равномерному распределению газа между обеими частями сосуда отвечает уже 20 возможных комбинаций молекул, т. е. 20 различных микросостояний:
| А | Б | Л | Б | А | Б | А | Б |
| 1,2,3 | 4,5,6 | 1,3,5 | 2,4,6 | 2,3,4 | 1,5,6 | 2,5,6 | 1,3,4 |
| 1,2,4 | 3,5,6 | 1,3,6 | 2,4,5 | 2,3,5 | 1,4,6 | 3,4,5 | 1,2,6 |
| 1,2,5 | 3,4,6 | 1,4,5 | 2,3,6 | 2,3,6 | 1,4,5 | 3,4,6 | 1,2,5 |
| 1,2,6 | 3,4,5 | 1,4,6 | 2,3,5 | 2,4,5 | 1,3,6 | 3,5,6 | 1,2,4 |
| 1,3,4 | 2,5,6 | 1,5,6 | 2,3,4 | 2,4,6 | 1,3,5 | 4,5,6 | 1,2,3 |
Теперь явление самопроизвольного сжатия газа, т е. сосредоточения всех его молекул в одной из частей сосуда, будет наблюдаться еще реже; равномерное же распределение газа по всему сосуду становится еще более вероятным.
Таким образом, с ростом числа молекул вероятность беспорядочного, равномерного распределения газа в сосуде очень быстро возрастает, а самопроизвольное сжатие газа становится все менее вероятным процессом. Если мы вспомним теперь, что макроскопические количества газа содержат огромное число молекул, то станет ясно, что в реальном опыте самопроизвольное сжатие газа представляет собой процесс практически невозможный, и что самопроизвольно будет протекать обратный процесс расширения газа, приводящий к равномерному, беспорядочному распределению его молекул по всему объему сосуда.
Рассмотренное нами явление расширения газа представляет собой пример проявления принципа направленности процессов к наиболее вероятному состоянию, т. е. к состоянию, которому соответствует максимальная беспорядочность распределения частиц. Направление самопроизвольного протекания химических реакций и определяется совокупным действием двух факторов: тенденцией к переходу системы в состояние с наименьшей внутренней энергией и тенденцией к достижению наиболее вероятного состояния.
Так, в приведенном примере с воздухом тенденция к минимуму потенциальной энергии заставляет молекулы, входящие в состав воздуха, падать на Землю, а тенденция к максимальной вероятности заставляет их беспорядочно распределяться в пространстве. В результате создается некоторое равновесное распределение молекул, характеризующееся более высокой их концентрацией у поверхности Земли и все большим разрежением по мере удаления от Земли.
В системах соль — вода минимум внутренней энергии в большинстве случаев соответствует кристаллическому состоянию соли. Однако наиболее вероятное состояние системы достигается при беспорядочном распределении соли в жидкой воде. В результате совместного действия этих двух факторов устанавливается равновесие, соответствующее определенной концентрации насыщенного раствора соли.
При химических реакциях в силу принципа направленности процессов к минимуму внутренней энергии атомы соединяются в такие молекулы, при образовании которых выделяется наибольшее количество энергии. В силу же принципа направленности процессов к наиболее вероятному состоянию протекают такие реакции, в ходе которых возрастает число частиц (например, реакции разложения молекул на атомы) или чисто возможных состояний атомов.
Так, в случае реакции
N2 + 3H2 = 2NH3+ 92,4 кДж
минимальной внутренней энергии системы соответствует аммиак, образующийся при протекании реакции до конца вправо. Однако наиболее вероятному состоянию системы отвечает азото-водород-ная смесь, образующаяся при полном разложении аммиака, ибо при этом в 2 раза возрастает число молекул газов. Вследствие действия обоих фактором в системе устанавливается равновесие, отвечающее определенному при данной температуре соотношению концентраций всех веществ. В случае реакции
N2 + O2 = 2NO - 180,8 кДж
минимальной внутренней энергии отвечает азото-кислородиая смесь, образующаяся при полном разложении оксида азота. Поскольку в ходе этой реакции число частиц не изменяется, то протекание реакции до конца как в прямом, так и в обратном направлении не увеличивает вероятности состояния системы. Не изменяется при этом и число возможных состояний атомов: в исходных веществах каждый атом и азота, и кислорода связан с атомом того же элемента (молекулы N2 и О2), а в продукте реакции каждый атом связан с атомом другого элемента (молекула N0). Иначе обстоит дело при частичном протекании процесса в прямом или в обратном направлении. В результате частичного прохождения реакции, т. е. при сосуществовании исходных веществ и продуктов реакции, атомы азота и кислорода находятся в двух состояниях: часть их связана в молекулы N2 и О2, а часть — в молекулы N0, Таким образом, число возможных микросостояний рассматриваемой системы, а следовательно, и вероятность соответствующего ее макросостояния возрастают при частичном протекании реакции. Итак, тенденция к уменьшению внутренней энергии способствует протеканию данной реакции до конца в обратном направлении, а тенденция к увеличению вероятности состояния вызывает ее частичное протекание в прямом направлении. Вследствие одновременного действия обоих факторов часть азото-кислородной смеси при нагревании превращается в N0 и устанавливается равновесие между исходными веществами и продуктом реакции.
Тенденция к переходу в состояние с наименьшей внутренней энергией проявляется при всех температурах в одинаковой степени. Тенденция же к достижению наиболее вероятного состояния проявляется тем сильнее, чем выше температура. Поэтому при низких температурах в большинстве случаев практически сказывается только влияние первой из этих тенденций, в результате чего самопроизвольно протекают экзотермические процессы. По мере возрастания температуры равновесие в химических системах все больше и больше сдвигается в сторону реакций разложения или увеличения числа состояний атомов. При этом каждой температуре отвечает состояние равновесия, характеризующееся определенным соотношением концентраций реагирующих веществ и продуктов реакции.
Оба рассмотренных фактора, а также результат их совместного действия можно выразить количественно. Величины, с помощью которых это делается, изучаются в разделе физики — термодинамике и называются термодинамическими величинами. К ним относятся, в частности, внутренняя энергия, энтальпия, энтропия и энергия Гиббса.
Термодинамические величины. Внутренняя энергия и энтальпия.
Внутренняя энергия U вещества (или системы) — это полная энергия частиц, составляющих данное вещество. Она слагается из кинетической и потенциальной энергий частиц. Кинетическая энергия — это энергия поступательного, колебательного и вращательного движения частиц; потенциальная энергия обусловлена силами притяжения и отталкивания, действующими между частицами.
Внутренняя энергия зависит от состояния вещества. Изменение внутренней энергии системы Δ U при том или ином процессе можно определять. Пусть в результате какого-нибудь процесса система переходит из начального состояния 1 в конечное состояние 2, совершая при этом работу А и поглощая из внешней среды теплоту Q. В термохимических уравнениях положительной принято считать теплоту, выделенную системой. В уравнениях термодинамики принято обратное условие: положительной считается теплота, поглощенная системой.
Внутренняя энергия системы уменьшится на величину А, возрастет на величину Q и в конечном состоянии будет равна
U2=Ul + Q - А
где U1 и U2— внутренняя энергия системы в начальном (1) и в конечном (2) состояниях. Если обозначить разность U2 — U1через Δ U, то уравнение можно представить в виде:
Δ U = Q - A
Это уравнение выражает закон сохранения энергии, согласно которому изменение внутренней энергии не зависит от способа проведения процесса, а определяется только начальным и конечным состояниями системы. Однако какая часть энергии пойдет на совершение работы, а какая превратится в теплоту — зависит от способа проведения процесса: соотношение между работой и теплотой может быть различным.
В частности, если в ходе процесса не производится никакой работы, в том числе работы расширения против внешнего давления, т. е., если объем системы не изменяется, то
Δ U = Qv
где Qv — теплота, поглощенная системой в условиях постоянного объема.
Последнее уравнение дает возможность определять изменение внутренней энергии при различных процессах. Например, в случае нагревания вещества при постоянном объеме изменение внутренней энергии определяется по теплоемкости этого вещества:
Δ U = Qv = nCv ΔT
Здесь Cv — молярная теплоемкость вещества при постоянном объеме; п — количество вещества; ΔT —разность между конечной и начальной температурами.
В случае химической реакции, протекающей без изменения объема системы, изменение внутренней энергии равно взятому с обратным знаком тепловому эффекту этой реакции.
Энтальпия.
Однако чаще в химии приходится иметь дело с процессами, протекающими при постоянном давлении. При этом удобно пользоваться величиной энтальпии Н, определяемой соотношением:
Н = U + PV
При постоянном давлении и при условии, что в ходе процесса совершается только работа расширения (А = PΔV). Работа (А)против силы внешнего давления равна
величине этой силы (F), умноженной на путь (Δ l), т. е. А = F Δl. Но сила равна давлению (P), (умноженному на ту площадь (S), на которую оно действует: F = PS, откуда А = PS Δ l или А = PΔV)
ΔН = ΔU + P ΔVили
ΔU = ΔН — Р ΔV
Сравнивая последнее уравнение с уравнением внутренней энергии
Δ U = Q - A
видим, что при указанных условиях ΔН = Qр, где где Qр— теплота, поглощенная системой при постоянном давлении.
Последнее уравнение дает возможность определять изменение энтальпии при различных процессах. Такие определения аналогичны определениям внутренней энергии, с той разницей, что все измерения должны проводиться в условиях постоянного давления. Так, при нагревании вещества изменение его энтальпии определяется по теплоемкости этого вещества при постоянном давлении
ΔН = QP = пСр ΔT
где п — количество вещества; Ср — молярная теплоемкость вещества при постоянном давлении.
При изменениях агрегатного состояния вещества и при аллотропных переходах изменение энтальпии равно по величине, но обратно по знаку теплоте соответствующего превращения (плавление, кипение, превращение из одной модификации в другую). Наконец, в случае химической реакции изменение энтальпии равно взятому с обратным знаком тепловому эффекту реакции, проведенной при постоянной температуре и постоянном давлении.
Энтальпия, как и внутренняя энергия, характеризует энергетическое состояние вещества, но включает энергию, затрачиваемую на преодоление внешнего
давления, т. е. на работу расширения. Подобно внутренней энергии, энтальпия определяется состоянием системы и не зависит от того, каким путем это состояние достигнуто. В случае газов различие между Δ U и ΔНв ходе того или
иного процесса может быть значительным. В случае систем, несодержащих газов, изменения внутренней энергии (Δ U) и энтальпии (ΔН), сопровождающие процесс, близки друг к другу. Это объясняется тем, что изменения объема ( Δ V) при
процессах,претерпеваемых веществами в конденсированных (т.е. в твердом или в жидком) состояниях, обычно очень невелики, и величина РΔV мала в сравнении с АН.
Термодинамические величины. Энтропия и энергия Гиббса.
Как уже говорилось, макросостояние системы тем более вероятно, чем большим числом микросостояний оно может осуществиться. Обычно число микросостояний, отвечающих тому или иному макросостоянию системы, очень велико. Это связано с тем, что в макроскопических количествах вещества число частиц колоссально велико, а их положения и скорости при обычных температурах чрезвычайно разнообразны.
Характеризовать в этом смысле состояние системы оказалось удобнее не самой вероятностью осуществления данного макросостояния, а величиной, пропорциональной ее логарифму. Эта величина называется энтропией. Энтропия (S) связана с числом (W) равновероятных микроскопических состояний, которыми можно реализовать данное макроскопическое состояние системы, уравнением
S = k lg W, где k — коэффициент пропорциональности
Наименьшую энтропию имеют идеально правильно построенные кристаллы при абсолютном нуле. Энтропия кристалла, в структуре которого имеются какие-либо неправильности, уже при абсолютном нуле несколько больше, так как нарушения идеальности могут реализоваться не единственным способом. С повышением температуры энтропия всегда возрастает, так как возрастает интенсивность движения частиц, а следовательно, растет число способов их расположения. Возрастает она также при превращении вещества из кристаллического состояния в жидкое и, в особенности, при переходе из жидкого состояния в газообразное. Изменяется энтропия и при протекании химических процессов. Эти изменения обычно особенно велики в случае реакций, приводящих к изменению числа молекул газов: увеличение числа газовых молекул приводит к возрастанию энтропии, уменьшение — к ее понижению.
Подобно внутренней энергии и энтальпии, энтропия зависит только от состояния системы. Но, в отличие от этих двух функций, связь изменения энтропии с теплотой зависит от способа проведения процесса — от его скорости.
Как уже говорилось, в ходе того или иного процесса соотношение между теплотой и производимой работой может быть различным. Только разность этих величин, равная изменению внутренней энергии системы, не зависит от способа осуществления процесса. При быстром его проведении работа бывает малой, а при медленном она возрастает. При бесконечно медленном осуществлении процесса — при проведении его бесконечно малыми шагами от одного состояния равновесия к следующему, бесконечно близкому к предыдущему, — работа принимает максимально возможное значение. Такое проведение процесса называется термодинамически обратимым, или просто обратимым (Термодинамическую обратимость как способ проведения процесса не следует путать с химической обратимостью - способностью реакции протекать и в прямом, и в обратном направлении).
В ряде случаев к обратимому проведению процесса можно приблизиться в экспериментальных условиях с высокой точностью. В лаборатории можно практически обратимо проводить окислительно-восстановительные реакции в гальванических элементах (см. § 98), плавление твердого тела, испарение жидкости. Если процесс проводится обратимо и при постоянной температуре (изотермически), то изменение энтропии связано с поглощаемой теплотой уравнением
ΔS = Qo6р/Т,
где Qo6p — количество теплоты, поглощенной системой в изотермическом обратимом процессе; Т— абсолютная температура.
С помощью этого уравнения можно определить, например, изменение энтропии при плавлении и кипении веществ.
Последнее уравнение показывает, что при поглощении некоторого количества теплоты энтропия системы возрастает тем сильнее, чем ниже температура, при которой поглощается теплота. Это можно пояснить следующим образом. Подведем одно и то же количество теплоты к двум одинаковым порциям данного вещества. При этом пусть одна из порции находится при низкой температуре, например 1 К, а другая — при высокой температуре, например 1000 К. Ясно, что относительное возрастание скорости движения частиц и увеличение степени их неупорядоченности, а следовательно, и возрастание энтропии в первом случае будет больше, чем во втором.
Энтропия имеет размерность энергии, деленной на температуру; выражают ее обычно в Дж/К.
Как показывается в термодинамике, можно ввести такие функции, которые отражают влияние на направление протекания процесса как тенденции к уменьшению внутренней энергии, так и тенденции к достижению наиболее вероятного состояния системы. Знак изменения подобной функции при той или иной реакции может служить критерием возможности самопроизвольного протекания реакции. Для изотермических реакций, протекающих при постоянном давлении, такой функцией является энергия Гиббс G, называемая также изобарно-изотермическим потенциалом, изобарным потенциалом или свободной энергией при постоянном давлении. Джозайя Уиллард Гиббс (1839—1903)—выдающийся американский физик, один из основателей химической термодинамики и статистической
физики.
Энергия Гиббса связана с энтальпией, энтропией и температурой соотношением:
G = H -TS
Если реакция осуществляется при постоянных давлении и температуре (такой процесс называется изобарно-изотермическим), то изменение энергии Гиббса при реакции будет равно:
ΔG = ΔН - TΔS
При обратимом и изотермическом проведении процесса AG равно по абсолютной величине, но обратно по знаку максимальной полезной работе, которую система производит в данном процессе:
ΔG = - Амакс
Полезной работой называется вся производимая в ходе процесса работа за вычетом работы расширения PΔV.
Можно показать, что в условиях постоянства температуры и давления реакции протекают самопроизвольно в сторону уменьшения энергии Гиббса. Поскольку ΔG равно по величине, но обратно по знаку максимальной полезной работе процесса, то сказанное можно сформулировать иначе: самопроизвольно могут протекать только те реакции, за счет энергии которых можно совершать полезную работу.
Для грубой оценки того, в каком направлении может протекать та или иная реакция при низких и при высоких температурах, можно воспользоваться приближенными уравнениями для изменения энергии Гиббса. При низких температурах множитель Т мал и абсолютное значение произведения TΔS тоже мало. В этом случае для реакций, имеющих значительный тепловой эффект, |ΔН|>>|TΔS|. Тогда в выражении
ΔG = ΔН - TΔS
вторым членом можно пренебречь. При этом получим
ΔG ≈ ΔН
При достаточно высоких температурах (множитель Т велик) имеем обратное соотношение:
|ΔН| << |TΔS|
Пренебрегая теперь первым членом в выражении энергии Гиббса, получим
ΔG = - TΔS
Эти приближенные равенства показывают, что при низких температурах критерием направления самопроизвольного протекания реакции в первом приближении может служить знак теплового эффекта реакции, а при высоких — знак изменения энтропии. Это означает, что при низких температурах самопроизвольно протекать могут экзотермические реакции, а при высоких — реакции, сопровождающиеся увеличением энтропии.
К сказанному необходимо добавить, что отрицательное значение ΔGтой или иной реакции указывает именно только на возможность ее протекания. В действительности реакция может при этом и не наблюдаться. Дело в том, что скорость ее может быть малой; тогда, несмотря на соблюдение условия ΔG< 0, реакция практически не будет протекать. В этих случаях для увеличения скорости реакции необходимо подобрать катализатор. Такое положение особенно часто наблюдается при низких температурах.
Дата добавления: 2015-08-21; просмотров: 409 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| Смещение химического равновесия. Принцип Ле Шателье. | | | Стандартные термодинамические величины. Химико-термодинамические расчеты. |