|
Читайте также: |
Р = CRT
Здесь Р — осмотическое давление раствора, кПа; С — его молярная концентрация (молярность), моль/л; R — универсальная газовая постоянная, 8,314 Дж/(моль-К); Т—абсолютная температура раствора.
Молярность раствора С представляет собой отношение количества растворенного вещества п к объему раствора V (л)
C = n/F
а количество вещества равно его массе т, деленной на молярную массу М. Отсюда для молярности раствора получаем:
С — tn/MV
Подставляя это значение С в уравнение Вант-Гоффа, найдем:
PV = mRT/M
Полученное уравнение по форме напоминает уравнение состояния идеального газа Клапейрона — Менделеева. Это уравнение позволяет по величине осмотического давления раствора определять молярную массу (а значит, и относительную молекулярную массу) растворенного вещества.
Пример. Осмотическое давление раствора, в 250 мл которого содержится 3 г сахара, при 12 °С равно 83,14 кПа. Определить относительную молекулярную массу сахара.
Подставляя данные в последнее уравнение, получаем
83,14 • 0,25 = 3-8,314 (273 + 12)/Af
откуда М = 342 г/моль. Относительная молекулярная масса сахара равна 342.
Если к раствору, отделенному от воды полупроницаемой перегородкой, приложить внешнее давление, равное осмотическому давлению раствора, то, как уже говорилось, осмос прекратится. Если же приложенное внешнее давление превысит осмотическое, то диффузия волы будет преимущественно происходить из раствора в водную фазу, т. е. в направленна, противоположном направлен вгао переноса воды при осмосе. Такое явление получило название обратного осмоса.
В настоящее время обратный осмос начали применять как один из най«' более экономичных способов опреснения воды. Солевой раствор (например, морскую воду) отделяют полупроницаемой мембраной от пресной воды и под[47] вергаюг давлению более высокому, чем осмотическое давление раствора. В ре- зультате часть содержащейся в растворе воды «вытесняется» в фазу пресной воды, а концентрация солей в оставшемся растворе повышается. Концентриро* ванный солевой раствор периодически заменяют свежими порциями подлежа» щей опреснению воды.
79. Давление пара растворов. При данной температуре давление насыщенного пара над каждой жидкостью — величина постоянная. Опыт показывает, что при растворении в жидкости какого» либо вещества давление насыщенного пара этой жидкости пони*, жается. Таким образом, давление насыщенного пара растворителя над раствором всегда ниже, чем над чистым растворителем при той же температуре. Разность между этими величинами принято называть понижением давления пара над раствором (или понижением давления пара раствора). Отношение величины этого понижения к давлению насыщенного пара над чистым раствори»: телем называется относительным понижением д а в л е «н и я пара над раствором.
Обозначим давление насыщенного пара растворителя над' чи«; стым растворителем через ро, а над раствором через р. Тогда относительное понижение давления пара над раствором будет представлять собою дробь:
(Ро — Р)/Ро
В 1887 г. французский физик Рауль, изучая растворы различных нелетучих * жидкостей и веществ в твердом состоянии, установил закон, связывающий понижение давления пара над разбавленными растворами неэлектролитов с концентрацией:
Относительное понижение давления насыщенного пара растворителя над раствором равно молярной доле растворенного вещества.
Математическим выражением закона Рауля является уравнен ние:
(Ро — Р)/Ро =
Здесь N2 — молярная доля растворенного вещества.
Явление понижения давления насыщенного пара над раствором вытекает из принципа Ле Шателье. Представим себе равновесие
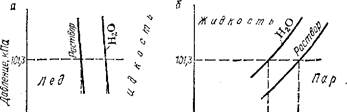
Рис. 73. Диаграмма состояния воды и водного раствора нелетучего вещества.
между жидкостью, например, водой, и ее паром. Это равновесие, которому отвечает определенное давление насыщенного пара, можно выразить уравнением
(Н20)жидк -<—^ (Н20)пар
Если теперь растворить в воде некоторое количество какого- либо вещества, то концентрация молекул воды в жидкости понизится и пойдет процесс, увеличивающий ее, — конденсация пара. Новое равновесие установится при более низком давлении насыщенного пара.
Понижение давления пара над раствором находит отражение на диаграмме состояния. На рис. 78 приведена схема диаграммы состояния воды и водного раствора нелетучего вещества. Согласно закону Рауля, давление водяного пара над водным раствором ниже, чем над водой. Поэтому кривая кипения для раствора лежит ниже, чем для воды. При переходе от воды к раствору изменяется также положение кривой плавления. И кривая кипения, и кривая плавления раствора расположены тем дальше от соответствующих кривых воды, чем концентрированнее раствор.
80. Замерзание и кипение растворов. Индивидуальные вещества характеризуются строго определенными температурами переходов из одного агрегатного состояния в другое (температура кипения, температура плавления или кристаллизации). Так, вода при нормальном атмосферном давлении (101,3 кПа) кристаллизуется при температуре 0 °С и кипит при 100 °С.
Иначе обстоит дело с растворами. Присутствие растворенного вещества повышает температуру кипения и понижает температуру замерзания растворителя, и тем сильнее, чем концентрированнее раствор. В большинстве случаев из раствора кристаллизуется (при замерзании) или выкипает (при кипении) только растворитель вследствие чего концентрация раствора в ходе его замерзания или кипения возрастает. Это, в свою очередь, приводит к еще большему повышению температуры кипения и снижению температуры замерзания. Таким образом, раствор кристаллизуется и кипит не при определенной температуре, а в некотором температурном интервале. Температуру начала кристаллизации и начала кипения данного раствора называют его температурой кристаллизации и температурой кипения.

|
Разность между температурами кипения раствора и чистого растворителя называют повышением температуры кипения раствора (Д£КцП). Разность между температурами замерзания чистого растворителя и раствора называют понижением
температуры замерзания раствора (А/зам). Обозначая температуры кипения п замерзания раствора /Кип и ^зам> а те же величины для чистого растворителя /,<ип п /зам, имеем:
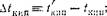
|
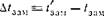
|
Всякая жидкость начинает кипеть при той температуре, при которой давление ее насыщенного пара достигает величины внешнего давления. Например, вода под давлением 101,3 кПа кипит при 100°С потому, что при этой температуре давление водяного пара как раз равно 101,3 кПа. Если же растворить в воде какое- нибудь нелетучее вещество, то давление ее пара понизится. Чтобы довести давление пара полученного раствора до 101,3 кПа, нужно нагреть раствор выше 100 °С. Отсюда следует, что температура кипения раствора всегда выше температуры кипения чистого растворителя. Аналогично объясняется и понижение температуры замерзания растворов.
Повышение температуры кипения и понижение температуры замерзания растворов соответствуют принципу Ле Шателье. Рассмотрим в этом плане замерзание раствора. Пусть имеется равновесие между жидкостью и твердой фазой, например, равновесие вода — лед при 0°С. Его можно выразить уравнением:
(Н2О)Т0 (Н20)жпдк
Если растворить в воде некоторое количество какого-либо вещества, то концентрация молекул воды в жидкости понизится и пойдет процесс, увеличивающий ее, — плавление льда. Для установления нового равновесия необходимо понизить температуру.
Повышение температуры кипения и понижение температуры замерзания находят отражение на диаграмме состояния. На рис. 79 приведена часть диаграммы состояния воды и раствора—отрезки кривых плавления и кипения в области давлений, близких
 i3mpacmtopa j I tsaM H?Q
i3mpacmtopa j I tsaM H?Q
|
| II % ! I. |
^иип 1 it//» оаствоон
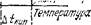
|
|
Рис. 79. Часть диаграммы состояния воды и раствора в области давлений, близких к 101,3 кПа (схема):
а —• кривые плавления; 0 — кривые кипения.
к нормальному атмосферному давлению (101,3 кПа). Отрезки пересечены горизонталью, отвечающей давлению 101,3 кПа (масштаб чертежа увеличен по сравнению с рис. 73 и 78). Видно, что точки пересечения этой горизонтали с.кривыми плавления и кипения для воды и для раствора различны. Абсциссы этих точек — температура замерзания и температура кипения — для воды равны 0 и 100 °С, а для раствора они соответственно ниже 0 °С и выше 100 °С. Кривые, отвечающие раствору, тем больше удалены от соответствующих кривых воды, чем концентрированнее раствор. Поэтому и разность между температурами кипения или замерзания воды и раствора тем больше, чем выше концентрация раствора.
Изучая замерзание и кипение растворов, Рауль установил, что для разбавленных растворов неэлектролитов повышение температуры кипения и понижение температуры замерзания пропорциональны концентрации раствора:
А^кип = Ет; Мзш = Km
Здесь т — молярная концентрация (моляльность); Е и К — эбуллиоскопическая[48] и криоскопическая[49] постоянные, зависящие только от природы растворителя, но не зависящие от природы растворенного вещества. Для воды криоскопическая постоянная К равна 1,86, эбуллиоскопическая постоянная Е равна 0,52. Для бензола К = 5,07, Е = 2,6.
На измерениях температур кипения и замерзания растворов основаны эбуллиоскопический и криоскопический методы определения молекулярных масс веществ. Оба метода широко используются в химии, так как, применяя различные растворители, можно определять молекулярные массы разнообразных веществ.
Пример. При растворении 2,76 г глицерина в 200 г воды температура замерзания понизилась на 0,279 градусов. Определить молекулярную массу глицерина.
Находим, сколько граммов глицерина приходится в растворе на 1000 г воды:
р = 2,76 • 1000/200 = 13,8 г
Выражаем моляльность раствора (т) через массу глицерина (р), приходящуюся на 1000 г воды, и его молярную массу (Af):
т = р/М = 13,8/М
Подставляем данные в уравнение:
Д/зам = /Сот; 0,279 = 1,86 • 13,8/М
Отсюда молярная масса глицерина М = 92 г/моль, а молекулярная масса равна 92.
Глава РАСТВОРЫ ЭЛЕКТРОЛИТОВ VIII
81. Особенности растворов солей, кислот и оснований. В главе VII мы познакомились с законами, которым подчиняются разбавленные растворы. Справедливость этих законов подтверждается результатами многих экспериментов. Однако имеются вещества, растворы которых сильно отклоняются от всех рассмотренных законов. К подобным веществам откосятся соли, кислоты и щелочи. Для них осмотическое давление, понижение давления пара, изме«нения температур кипения и замерзания всегда больше, чем ето отвечает концентрации раствора.
Например, понижение температуры замерзания раствора, содержащего 1 г NaCl в 100 г воды, почти вдвое превышает Д^зам, вычисленное по закону Рауля. Во столько же раз и осмотическое давление этого раствора больше теоретической величины.
Как указывалось в § 78, величина осмотического давления выражается уравнением:
Р = CRT
Чтобы распространить это уравнение на растворы с «ненормальным» осмотическим давлением, Ванг-Гофф ввел в него поправочный коэффициент i (изотонический[50] коэффициент)1, показывающий, во сколько раз осмотическое давление данного раствора больше «нормального»:
Р = iCRT
Коэффициент i определялся для каждого раствора экспериментальным путем — например, по понижению давления пара, или по понижению температуры замерзания, или по повышению температуры кипения.
Обозначим через Р' осмотическое давление раствора, через Мяш — повышение температуры кипения, Д^зам — понижение температуры замерзания раствора, не подчиняющегося законам Вант- Гоффа и Рауля, а через Р, Д^кип и Д?3ая — значения тех же величин, вычисленные теоретически по концентрации раствора. Поскольку и осмотическое давление, и изменения температур замерзания и кипения пропорциональны числу находящихся в растворе частиц растворенного вещества, то коэффициент i можно выразить отношениями:
= Р1Р = Л'кипМиП = Д'зам/Д'эли
Значения коэффициента £, найденные Вант-Гоффом для 0,2 н.
растворов некоторых солей по понижению их температур замерзания, проведены в табл. 11.
Таблица 11. Значение коэффициента / для 0,2 н. растворов некоторых солей
|
Данные табл. 11 показывают, что коэффициент i для различных солей различен. С разбавлением раствора он растет, приближаясь к целым числам 2, 3, 4. Для солей аналогичного состава эти числа одинаковы. Например, для всех солей, образованных одновалентными металлами и одноосновными кислотами, при достаточном разбавлении их растворов коэффициент i приближается к 2; для солей, образованных двухвалентными металлами и одноосновными кислотами,—ж 3.
Итак, соли, кислоты и основания растворяясь в воде, создают значительно большее осмотическое давление, чем эквимолекулярные количества всех остальных веществ. Как же объяснить это явление?
Отметим, что аналогичное явление наблюдаете.! г отношении некоторых газов или веществ, переходящих в газе образное состояние. Например, пары пентахлорида фосфора PClg, иода и некоторых других веществ при нагревании в закрытом сосуде обнаруживают более высокое давление, чем следует по закону Гей- Люссака.
Для газов это явление объясняется диссоциацией. Если, например, PCI5 полностью разложится на РС13 и С12, то понятно, что при неизменном объеме давление, зависящее от числа частиц, должно увеличиться вдвое. При неполной диссоциации, когда только часть молекул подверглась разложению, давление также возрастает, но менее, чем вдвое.
Естественно было предположить, что в растворах, обладающих ненормально высоким осмотическим давлением, молекулы растворенного вещества тоже распадаются на какие-то более мелкие частицы, так что общее число частиц в растворе возрастает. А поскольку осмотическое давление зависит от числа частиц растворенного вещества, находящихся в единице объема раствора, то с увеличением этого числа оно тоже увеличивается. Такое предположение впервые было высказано в 1887 г. шведским ученым
Аррениусом [51] и легло в основу его теории, объясняющей поведение солей, кислот и оснований в водных растворах.
Водные растворы солей, кислот и оснований обладают еще одной особенностью — они проводят электрический ток. При этом большинство твердых солей и оснований в безводном состоянии, а также безводные кислоты обладают очень слабой электрической проводимостью; плохо проводит электрический ток и вода. Очевидно, что при образовании растворов подобные вещества претерпевают какие-то изменения, обусловливающие возникновение высокой электрической проводимости. Как мы увидим ниже, эти изменения заключаются в диссоциации, соответствующих веществ на ионы, которые и служат переносчиками электрического тока.
Вещества, проводящие электрический ток своими нонами, называются электролитами. При растворении в воде и в ряде неводных растворителей свойства электролитов проявляют соли, кислоты н основания. Электролитами являются также многие расплавленные соли, оксиды и гидрокспды, а также некоторые соли и оксиды в твердом состоянии.
82. Теория электролитической диссоциации. Аррениус обратил внимание на тесную связь между способностью растворов солей, кислот и оснований проводить электрический ток и отклонениями растворов этих веществ от законов Вант-Гоффа и Рауля. Он показал, что по электрической проводимости раствора можно рассчитать его осмотическое давление, а следовательно, и поправочный коэффициент i. Значения г, вычисленные им из электрической проводимости, хорошо совпали с-величинами, найденными для тех же растворов иными методами.
Причиной чрезмерно высокого осмотического давления растворов электролитов является, согласно Аррениусу, диссоциация электролитов на ионы. Вследствие этого, с одной стороны, увеличивается общее число частиц в растворе, а следовательно, возрастают осмотическое давление, понижение давления пара и изменения темцератур кипения и замерзания, с другой — ионы обусловливают способность раствора проводить электрический ток.
Эти предположения в дальнейшем были развиты в стройную теорию, получившую название теории электролитической диссоциации. Согласно этой теории, при растворении в воде электролиты распадаются (диссоциируют) на положительно и отрицательно заряженные ионы. Положительно заряженные ионы называются катионами; к ним относятся, например, ионы водорода и металлов. Отрицательно заряженные ионы называются анионами; к ним принадлежат ионы кислотных остатков и гидроксид-ионы. Как и молекулы растворителя, ионы в растворе находятся в состоянии неупорядоченного теплового движения.
Процесс электролитической диссоциации изображают, пользуясь химическими уравнениями. Например, диссоциация НС1 выразится уравнением:
НС1 = Н+ + СГ
Распад электролитов на ионы объясняет отклонения от законов Вант-Гоффа и Рауля, о которых говорилось в начале этой главы. В качестве примера мы приводили понижение температуры замерзания раствора NaCl. Теперь нетрудно понять, почему понижение температуры замерзания этого раствора столь велико. Хлорид натрия переходит в раствор в виде ионов Na+ н СР. При этом из одного моля NaCl получается не 6,02 ■ 1023 частиц, а вдвое большее их число. Поэтому и понижение температуры замерзания в растворе NaCl должно быть вдвое больше, чем в растворе неэлектролита той же концентрации.
Точно так же в очень разбавленном растворе хлорида бария, диссоциирующего согласно уравнению
ВаС12= Ва2+ +га-
осмотическое давление оказывается в 3 раза больше, чем вычисленное по закону Вант-Гоффа, так как число частиц в растворе в 3 раза больше, чем если бы хлорид бария находился в нем в виде молекул ВаС12.
Таким образом, особенности водных растворов электролитов, противоречащие с первого взгляда законам Вант-Гоффа и Рауля, были объяснены на основе этих же законов.
Однако теория Аррениуса не учитывала всей сложности явлений в растворах. В частности, она рассматривала ионы как свободные, независимые от молекул растворителя частицы. Теории Аррениуса противостояла химическая, или гидратная, теория растворов Менделеева, в основе которой лежало представление о взаимодействии растворенного вещества с растворителем. В преодолении кажущегося противоречия обеих теорий большая заслуга принадлежит русскому ученому И, А. Каблукову [52], впервые высказавшему предположение о гидратации ионов. Развитие этой идеи привело в дальнейшем к объединению теорий Аррениуса и Менделеева.
83. Процесс диссоциации. В зависимости от структуры растворяющегося вещества в безводном состоянии его диссоциация протекает по-разному. Наиболее типичны при этом два случая. Один из них — это диссоциация растворяющихся солей, т. е. кристаллов
 Pile. 80. Схема растворения соли.
Pile. 80. Схема растворения соли.
|
с ионной структурой, второй — диссоциация при растворении кислот, т. е. веществ, состоящих из полярных молекул.
Когда кристалл соли, например, хлорида калия, попадает в воду, то расположенные на его поверхности ионы притягивают к себе полярные молекулы воды (ион-дипольное взаимодействие). К ионам калия молекулы воды притягиваются своими отрицательными полюсами, а к хлорид-ионам — положительными (рис. 80). Но, если ионы притягивают к себе молекулы воды, то и молекулы воды с такой же силой притягивают к себе ионы. В то же время притянутые молекулы воды испытывают толчки со стороны других молекул, находящихся в движении. Этих толчков вместе с тепловыми колебаниями ионов в кристалле оказывается достаточно для отделения ионов от кристалла и перехода их в раствор. Вслед за первым слоем ионов в раствор переходит следующий слой, и таким образом идет постепенное растворение кристалла.
Иначе протекает диссоциация полярных молекул (рис. 81). Молекулы воды, притянувшиеся к концам полярной молекулы (ди- поль-дипольное взаимодействие), вызывают расхождение ее полюсов— поляризуют молекулу. Такая поляризация в сочетании с колебательным тепловым движением атомов в рассматриваемой молекуле, а также с непрерывным тепловым движением окружающих ее молекул воды приводит в конечном счете к распаду полярной молекулы на ионы. Как и в случае растворения ионного кристалла,
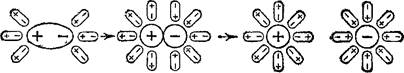 Рис. 81. Схема диссоциации полярных молекул в растворе.
Рис. 81. Схема диссоциации полярных молекул в растворе.
|
эти ионы гидратируютея. При этом ион водорода Н+ (т. е. протон); оказывается прочно связанным с молекулой воды в ион гидро- ксония Н30+. Так, при растворении в воде хлороводорода происходит процесс, который схематически можно выразить уравнением:
Н20 + НС! = Н30+ С Г
В результате этого процесса молекула НС1 расщепляется таким образом, что общая пара электронов остается у атома хлора, который превращается в ион С1~, а протон, внедряясь в электронную оболочку атома кислорода в молекуле воды, образует нон гидро- ксония Н30+.
Подобного же рода процессы происходят и при растворении в воде других кислот, например, азотной:
Н20 + НХ03 = Н30+ + NO3
Перешедшие в раствор ноны остаются связанными с молекулами воды и образуют гидраты ионов. Иначе говоря, в результате диссоциации образуются не свободные ионы, а соединения ионов с молекулами растворителя. В общем случае любого растворителя эти соединения называются сольватами ионов. Но в уравнениях диссоциации обычно пишут формулы ионов, а не их гидратов или сольватов, тем более что число молекул растворителя, связанных с ионами, изменяется в зависимости от концентрации раствора и других условий.
Диссоциации веществ как ионного, так и молекулярного строения способствует полярность молекул растворителя. Поэтому не только вода, но и другие жидкости, состоящие из полярных молекул (муравьиная кислота, этиловый спирт, аммиак и другие), также являются ионизирующими растворителями: соли, кислоты и основания, растворенные в этих жидкостях, диссоциируют на ионы.
84. Степень диссоциации. Сила электролитов. Если бы электролиты полностью диссоциировали на ионы, то осмотическое давление (и другие пропорциональные ему величина) всегда было бы в целое число раз больше значений, наблюдаемых в растворах неэлектролитов. Но еще Вант-Гофф установил, что коэффициент / выражается дробными числами, которые с разбавлением раствора возрастают, приближаясь к целым числам.
Аррениус объяснил этот факт тем, что лишь часть электролита диссоциирует в растворе на ионы, и ввел понятие степени диссоциации. Степенью диссоциации электролита называется отношение числа его молекул, распавшихся в данном растворе на ионы, к общему числу его молекул в растворе.
Позже было установлено, что электролиты можно разделить на две группы: сильные и слабые электролиты. Сильные электролиты в водных растворах диссоциированы практически нацело. Понятие степени диссоциации к ним по существу неприменимо, а отклонение изотонического коэффициента i от целочисленных значений объясняется другими причинами (см. § 86). Слабые электролиты в водных растворах диссоциируют только частично, и в растворе устанавливается динамическое равновесие между недиссоцииро- ванными молекулами и нонами.
К сильным электролитам принадлежат почти все соли; из важнейших кислот и оснований к ним относятся hn03, h2s04, НСЮ4, НС1, НВг, HI, КОН, NaOH, Ва(ОН)2 и Са(ОН)2,
К слабым электролитам относится большинство органических кислот, а из важнейших неорганических соединений к ним принадлежат Н2СОз, H2s, HCN, H2Si03 и nh4oi-i.
Степень диссоциации принято обозначать греческой буквой а и выражать либо в долях единицы, либо в процентах. Так, для 0,1 н. раствора СН3СООН а ==0,013 (или 1,3%), а для 0,1 н. раствора HCN а = Ю-4 (или 0,01 %).
85. Константа диссоциации. К равновесию, которое устанавливается в растворе слабого электролита между молекулами и ионами, можно применить законы химического равновесия и записать выражение константы равновесия. Например, для диссоциации уксусной кислоты
СНзСООН Н+ СНзСОСГ
константа равновесия имеет вид
[Н+] [СНзСОСГ]
К-
[СНзСООН]
Здесь в числителе дроби стоят концентрации ионов —• продуктов диссоциации, а в знаменателе — концентрация недиссоцииро- ванных молекул.
Константа равновесия, отвечающая диссоциации слабого электролита, называется константой диссоциации. Величина К зависит от природы электролита и растворителя, а также от температуры, но не зависит от концентрации раствора. Она характер ризует способность данной кислоты или данного основания распадаться ца ионы: чем выше К, тем легче электролит диссоциирует* Многоосновные кислоты, а также основания двух- и более валентных металлов диссоциируют ступенчато. В растворах эти}8 веществ устанавливаются сложные равновесия, в которых участ-* вуют ионы различного заряда. Например, диссоциация угольной кислоты происходит в две ступени:
Н2СОэ H+ + HCOJ
НСОз н+ + со
Первое равновесие —д и с с о ц и а ц и я по первой ступе' и и — характеризуется константой диссоциации, обозначаемой Kit
К,. [н+3 [НСО-]
а второе — диссоциация по второй ступени — константой диссоциации -Кг:
к __ [Н1[С0Г]
[нсо;]
Суммарному равновесию
Н2С03 2Н+ + СОз"
отвечает суммарная константа диссоциации К:
у [Hi [cor]
1НгСОз]
Величины К, К\ и Кг связаны друг с другом соотношением:
К = /С,К»
Аналогичные соотношения характеризуют и ступенчатую диссоциацию оснований многовалентных металлов. Например, двум ступеням диссоциации гидроксида железа(II)
Fe(OH)2 =?=*= FeOH+ + OH~
FeOH+ Fei+ + OH~
отвечают константы диссоциации:
,, [геОН+] [ОН-] „ [Fea1 [ОН~]
l\i— ------------------------------- И Д2 = ---------;---------- —----
[Fe(OH)2] [FeOH+]
Сум м арной диссо ци а цн и
Fe(OH)a Fe2++ 20Н-
отвечает константа:
iFe'lIOH-]»
[Fe(OH)2]
При этом
к = к,к2
При ступенчатой диссоциации веществ распад по последующей ступени всегда происходит в меньшей степени, чем по предыдущей (по второй меньше, чем по первой и т. д.). Иначе говоря, соблюдается неравенство:
К^ЖгЖг...
Это объясняется тем, что энергия, которую нужно затратить для отрыва иона, минимальна при его отрыве от нейтральной молекулы и становится больше при диссоциации по каждой следующей ступени.
Если обозначить концентрацию электролита, распадающегося на два иона, через С, а степень его диссоциации в данном растворе через а, то концентрация каждого из ионов будет Са, а концентрация недиссоциированных молекул С(1—а). Тогда уравне-
| -С |
| К = |
| 1 — а |
ние константы диссоциации принимает вид (Са)2
Дата добавления: 2015-08-21; просмотров: 120 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| Глава ВОДА, VII РАСТВОРЫ 2 страница | | | Глава ВОДА, VII РАСТВОРЫ 4 страница |