Читайте также:
|
Безопасное и эффективное применение лекарственных средств предусматривает доставку их к тканям-мишеням в таких находящихся в достаточно узком диапазоне концентрациях, которые обеспечивали бы эффективность действия без проявлений токсичности. Это обеспечивается соблюдением режимов его введения, основанных на кинетических свойствах данного лекарственного средства и механизмах его доставки к мишеням. В этой главе изложены принципы выведения из организма и распределения лекарственного средства в органах и тканях, лежащие в основе оптимальных режимов введения больному нагрузочных и поддерживающих доз данного препарата, и рассмотрены случаи нарушения выведения лекарственного средства из организма (например, при почечной недостаточности). Уделено внимание также кинетическим основам оптимального использования данных об уровне содержания лекарственных средств в плазме крови.
Содержание лекарственного препарата в плазме крови после введения единичной дозы. Уменьшение уровня содержания лидокаина в плазме крови после его внутривенного введения, как показано на рис. 64-1, имеет двухфазный характер; такое снижение концентрации типично для многих лекарственных средств. Сразу после быстрого введения в организм по существу весь лекарственный препарат находится в плазме крови, а затем переносится в ткани, и отрезок времени, в течение которого происходит этот перенос, называется фазой распределения. Для лидокаина она составляет 30 мин, после чего происходит медленное снижение уровня содержания препарата, называемое фазой уравновешивания, или выведения, в течение которой уровни содержания лекарственного препарата в плазме крови и тканях находятся в псевдоравновесии.
Фаза распределения. Процессы, происходящие во время фазы распределения, зависят от того, будет ли уровень содержания лекарственного средства в месте локализации его рецептора близок к уровню его содержания в плазме крови. Если это условие будет соблюдено, то фармакологическое действие препарата в этот период (благоприятное либо неблагоприятное) может оказаться чрезмерным. Например, после введения небольшой дозы (50 мг) лидокаина его противоаритмическое действие проявится в раннем периоде фазы распределения, но прекратится, как только уровень содержания лидокаина упадет ниже минимально эффективного, даже в том случае, если равновесие между уровнями его содержания в плазме крови и в тканях достигнуто не будет. Таким образом, для достижения такого эффекта, который бы поддерживался и во время фазы уравновешивания, следует вводить большую единичную дозу или несколько маленьких доз. Однако токсичность высоких концентраций некоторых лекарственных средств, проявляющаяся во время фазы распределения, препятствует внутривенному введению такой единичной нагрузочной дозы, которая обеспечивала бы терапевтический уровень содержания препарата в течение фазы уравновешивания. Например, введение нагрузочной дозы фенитоина в виде однократной внутривенной инъекции может вызвать сердечно-сосудистый коллапс, обусловленный высокими уровнями содержания фенитоина во время фазы распределения. Если нагрузочная доза фенитоина вводится внутривенно, это следует делать дробно, с интервалами, достаточными для распределения предыдущей дозы препарата, прежде чем будет введена последующая (например, по 100 мг через каждые 3—5 мин). По этим же причинам нагрузочную дозу при внутривенном введении многих сильнодействующих лекарственных средств, быстро достигающих равновесных концентраций в местах локализации их рецепторов, вводят по частям.
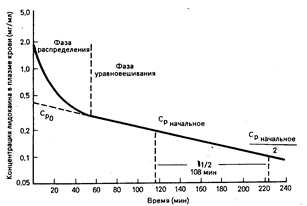
Рис. 64-1. Концентрации лидокаина в плазме крови после внутривенного введения 50 мг препарата.
Время полувыведения (108 мин) — это время, необходимое для уменьшения любого заданного уровня содержания лидокаина во время фазы уравновешивания (Срначальное) до,половины первоначального значения. Ср0 представляет собой гипотетическое значение концентрации лидокаина в плазме крови в точке времени 0 в том случае, если бы равновесное состояние было достигнуто мгновенно.
При пероральном введении единичной дозы препарата, обеспечивающей попадание в систему кровообращения эквивалентного количества лекарственного средства, уровни его содержания в плазме крови во время фазы распределения возрастают не так резко, как после внутривенного введения. Поскольку абсорбция лекарственного препарата после перорального введения происходит постепенно и он попадает в систему кровообращения достаточно медленно, распределение большей части препарата будет окончено к тому времени, когда завершится его абсорбция. Так, новокаинамид, который почти полностью абсорбируется после перорального введения, можно вводить перорально в виде единичной нагрузочной дозы, равной 750 мг, почти не рискуя вызвать развитие гипотонии; тогда как внутривенно эту дозу препарата безопаснее вводить частями, примерно по 100 мг каждая, с интервалом в 5 мин, чтобы предотвратить развитие гипотонии во время фазы распределения в случае единовременного введения всей нагрузочной дозы.
Другие лекарственные средства поступают к местам их фармакологического действия во время фазы распределения медленно. Например, уровень содержания дигоксина в месте локализации его рецепторов (и его фармакологического действия) не соответствует уровню его содержания в плазме крови во время фазы распределения. Дигоксин транспортируется к своим сердечным рецепторам (или связывается с ними) на протяжении всей фазы распределения. Таким образом, уровень его содержания в плазме крови снижается в течение фазы распределения, длящейся несколько часов, в то время как уровень содержания в месте его действия и фармакологического эффекта возрастает. Только к концу фазы распределения, когда будет достигнуто равновесие уровней содержания дигоксина в плазме крови и на месте локализации рецепторов, концентрация препарата в плазме крови будет действительно отражать его фармакологический эффект. Должно пройти яе менее 6—8 ч, пока не закончится фаза распределения и можно будет ориентироваться на концентрацию дигоксина в плазме крови в качестве реального показателя для оценки лечебного эффекта.
Фаза уравновешивания. После того как распределение завершается достижением равновесия концентраций лекарственного препарата в плазме крови и в тканях, уровни содержания его начинают снижаться с одинаковой скоростью по мере того, как препарат выводится из организма. Поэтому фазу уравновешивания иногда называют также фазой выведения.
Выведение большинства лекарственных средств происходит как процесс первого порядка. Для процесса первого порядка во время фазы уравновешивания характерно то, что время, необходимое для уменьшения уровня содержания лекарственного препарата в плазме крови до половины его начального значения (период полувыведения, ti/,), одинаково независимо от того, какая именно точка на кривой изменения концентрации препарата в плазме крови будет выбрана в качестве начальной точки для выполнения измерения. Другой характерной чертой процесса первого порядка во время фазы уравновешивания является линейная зависимость величины концентрации препарата в плазме крови от времени на полулогарифмическом графике. Из графика, отражающего снижение концентрации лидокаина (см. рис. 64-1), видно, что время его полувыведения составляет 108 мин.
Несложно рассчитать, какая доля препарата из введенной дозы остается к организме в любой прошедший после введения отрезок времени, выраженный через число периодов полувыведения:
| Число периодов полувыведения | Часть дозы, остающейся в организме (%) |
| 12,5 | |
| 6,25 | |
| 3,125 |
Теоретически процесс выведения никогда не завершается полностью. Однако с клинической точки зрения выведение можно считать завершенным после того, как будет выведено 90% введенной дозы. Поэтому в практике процесс выведения первого порядка считают завершенным по истечении 3— 4 периодов полувыведения.
Накопление лекарственного средства — нагрузочная и поддерживающая дозы. При повторном введении лекарственного средства количество его в организме будет накапливаться, если выведение первой дозы не завершено до введения второй, и как количество препарата в организме, так и его фармакологическое действие будут возрастать в случае продолжающегося введения до тех пор, пока их значения не достигнут плато. Накопление в организме дигоксина, вводимого в повторных поддерживающих дозах (без нагрузочной дозы), проиллюстрировано на рис. 64-2. Так как период полувыведения дигоксина составляет приблизительно 1,6 сут у больного с нормальной функцией почек, то к концу первых суток в организме останется 65% введенной дозы препарата. Таким образом, вторая доза увеличит количество дигоксина в организме (и средний уровень его содержания в плазме крови) до 165% того количества, которое осталось в организме после введения первой дозы. Каждая последующая доза будет приводить к накоплению все большего количества препарата в организме до тех пор, пока не будет достигнуто плато. При достижении плато, устойчивого состояния, в единицу времени в организм
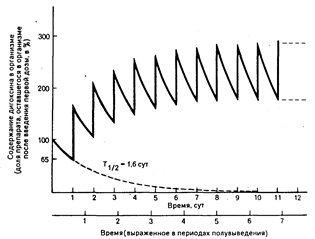
Рис. 64-2. Накопление дигоксина во времени при введении единичной ежедневной поддерживающей дозы в отсутствие нагрузочной дозы.
Независимо от величины нагрузочной дозы после проведения поддерживающей терапии в течение времени, соответствующего 3—4 периодам полувыведения, количество лекарственного средства в организме определяется величиной поддерживающей дозы. Независимость уровней содержания лекарственного средства в плазме крови при устойчивом состоянии от его нагрузочной дозы проиллюстрировано на рис. 64-3, из которого видно, что выведение любого лекарственного средства практически завершается через 3—4 периода его полувыведения.
Факторы, определяющие уровни содержания лекарственного средства в плазме крови в течение фазы уравновешивания. Важным фактором, определяющим уровень содержания лекарственного средства в плазме крови в течение фазы уравновешивания после введения единичной дозы, является степень распределения его в организме. Например, если распределение дозы в 3 мг крупномолекулярного лекарственного средства ограничивается объемом плазмы крови, равным 3 л, то его концентрация в плазме будет составлять 1 мг/л. Однако если лекарственное средство распределяется таким образом, что 90% его количества покидает плазму, то в 3 литрах ее объема останется только 0,3 мг, и концентрация этого лекарственного средства в плазме крови будет составлять 0,1 мг/л. Степень внесосудистого распределения в фазе уравновешивания может быть выражена кажущимся объемом распределения, или Vd, выражающим зависимость между количеством лекарственного средства в организме и его концентрацией в плазме крови в фазе уравновешивания:

Количество лекарственного средства в организме выражается в единицах массы (например, в миллиграммах), а его концентрация в плазме крови—в единицах массы, приходящейся на единицу объема (например, в миллиграммах на литр). Таким образом, Vd—это гипотетический объем, в котором было бы распределено некоторое количество лекарственного средства, если бы его концентрация во всем этом объеме была равна его концентрации в плазме крови. Хотя эта величина не отражает реальный объем, она представляется важной, поскольку определяет долю общего количества лекарственного средства, содержащегося в плазме крови, а следовательно, и ту его долю, которая будет выведена из организма. Приближенное значение Vd в фазе уравновешивания можно получить путем определения концентрации лекарственного средства в плазме крови во временной точке 0 (Ср0) при помощи обратной экстраполяции кривой фазы уравновешивания к временной точке 0 (см. рис. 64-1). Сразу после внутривенного введения лекарственного средства, когда его количество в организме во временной точке будет равно введенной дозе:
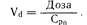
При введении крупномолекулярного лекарственного средства, упомянутого выше, величина Ср0 в 1 мг/л после введения дозы 3 мг, согласно формуле, указывает на то, что Vd — это реальный объем, равный объему плазмы крови. Однако этот случай является исключением, так как для большинства лекарственных средств величина Vd будет больше, чем объем плазмы крови; поглощение многих лекарственных средств клетками настолько значительно, что уровни их содержания в тканях превышают соответствующие значения в плазме крови. Для таких лекарственных средств гипотетическая величина Vd велика и превышает объем всей жидкости в организме. Например, величина Ср0, полученная путем экстраполяции после введения 50 мг лидоксина, составляет 0,42 мг/л, откуда следует, что величина Vd равна 119 л (см. рис. 64-1).
Поскольку выведение лекарственных средств из организма осуществляется главным образом почками и печенью, целесообразно рассматривать этот вопрос в соответствии с понятием клиренса. Например, в почках независимо от того, в какой степени выведение лекарственного препарата обусловливается фильтрацией, секрецией или реабсорбцией, конечным результатом является уменьшение концентрации лекарственного средства в плазме крови по мере его прохождения через этот орган. Степень уменьшения концентрации лекарственного средства выражается в виде коэффициента экстракции, или Е, который является постоянной величиной в течение всего времени, пока происходит выведение как процесс первого порядка:
 где Са — концентрация в плазме артериальной крови; Св — концентрация в плазме венозной крови.
где Са — концентрация в плазме артериальной крови; Св — концентрация в плазме венозной крови.
Если экстракция завершена, то Е= 1. Если суммарный протекающий через почки в единицу времени поток равен Q (мл/мин), то общий объем плазмы, из которого полностью удаляется лекарственное средство в единицу времени (клиренс из организма, С1), определяется как Спочек = QE.
Если коэффициент почечной экстракции пенициллина равен 0,5, а поток плазмы через почки составляет 680 мл/мин, то почечный клиренс пенициллина будет равен 340 мл/мин. Если коэффициент экстракции высок, как в случае почечной экстракции аминогиппурата или печеночной экстракции пропранолола, то клиренс будет являться функцией кровотока в данном органе.
Клиренс лекарственного средства из организма — сумма клиренсов из всех органов выведения — служит самой лучшей мерой эффективности процессов выведения. Если лекарственное средство выводится и почками, и печенью, то:
Cl=Clпочек + Сlпечени
Таким образом, если у здорового человека пенициллин выводится посредством почечного клиренса, равного 340 мл/мин, и печеночного, равного 36 мл/мин, то общий клиренс составит 376 мл/мин. Если почечный клиренс снизится наполовину, то величина общего клиренса составит 170-1-36, или-206 мл/мин. При анурии общий клиренс будет равен печеночному клиренсу.
Во время каждого прохождения крови через орган выведения из организма может быть удалена только та доля лекарственного средства, которая находится в плазме крови. Чтобы установить влияние клиренса плазмы одним или несколькими органами на скорость выведения лекарственного препарата из организма, необходимо связать клиренс с объемом «плазменных эквивалентов», подлежащих очищению, т. е. с объемом распределения. Если объем распределения составляет 10000 мл, а клиренс равен 1000 мл/мин, то за 1 мин будет выводиться 1/10 доля всего количества лекарственного средства, находящегося в организме. Эту величину, Cl/Vd, называют константой долевой скорости выведения и обозначают символом k:

Умножив величину k на общее количество лекарственного средства, находящегося в организме, можно определить фактическую скорость выведения в любой данный момент времени:

Это общее для всех процессов первого порядка уравнение гласит, что скорость выведения вещества пропорциональна уменьшению его количества.
Поскольку период полувыведения t1/2 является временным выражением экспоненциального процесса первого порядка, он связан с константой долевой скорости выведения k следующим образом:

Если лекарственное средство присутствует в форменных элементах крови, расчет его экстракции и клиренса из крови более физиологичен, чем из плазмы; поскольку
 то
то

Линейная зависимость между k и клиренсом креатинина дает возможность использовать k для расчета изменений в выведении лекарственного средства при уменьшении клиренса креатинина в случае почечной недостаточности. Период полувыведения связан с величиной клиренса нелинейной зависимостью. Зависимость
 отражает влияние клиренса и объема распределения на период полувыведения. Так, период полувыведения укорачивается при стимулировании фенобарбиталом активности ферментов, ответственных за печеночный клиренс лекарственного средства, и удлиняется, если почечный клиренс лекарственного средства снижается вследствие почечной недостаточности. Кроме того, укорочению периода полувыведения некоторых лекарственных средств способствует уменьшение объема их распределения. Так, например, если при сердечной недостаточности объем распределения уменьшается параллельно с уменьшением клиренса, снижение клиренса вызовет лишь очень небольшие изменения в продолжительности периода полувыведения лекарственного препарата, но уровень его содержания в плазме крови возрастет, как это происходит в случае с лидокаином. При лечении больных после передозировки лекарственных средств влияние гемодиализа на их выведение будет зависеть от объема распространения. Если объем распространения велик, как в случае трициклических антидепрессантов [Vd дезипрамина (Desipramine) превышает 2000 л], выведение такого препарата, даже с помощью диализатора с высоким клиренсом, будет происходить медленно.
отражает влияние клиренса и объема распределения на период полувыведения. Так, период полувыведения укорачивается при стимулировании фенобарбиталом активности ферментов, ответственных за печеночный клиренс лекарственного средства, и удлиняется, если почечный клиренс лекарственного средства снижается вследствие почечной недостаточности. Кроме того, укорочению периода полувыведения некоторых лекарственных средств способствует уменьшение объема их распределения. Так, например, если при сердечной недостаточности объем распределения уменьшается параллельно с уменьшением клиренса, снижение клиренса вызовет лишь очень небольшие изменения в продолжительности периода полувыведения лекарственного препарата, но уровень его содержания в плазме крови возрастет, как это происходит в случае с лидокаином. При лечении больных после передозировки лекарственных средств влияние гемодиализа на их выведение будет зависеть от объема распространения. Если объем распространения велик, как в случае трициклических антидепрессантов [Vd дезипрамина (Desipramine) превышает 2000 л], выведение такого препарата, даже с помощью диализатора с высоким клиренсом, будет происходить медленно.
Величина доли лекарственного вещества, экстракция которой обеспечивается органами выведения, также определяется степенью связывания лекарственного средства с белками плазмы крови. Однако изменение степени связывания с белками будет значительно влиять на коэффициент экстракции только в тех случаях, когда выведение ограничивается не связанной с белками (свободной) фракцией лекарственного вещества в плазме. Степень влияния связывания лекарственного средства с белком на выведение зависит от его относительного сродства к процессу связывания с белками плазмы и к процессу выведения. Так, высокая степень сродства транспортной анионной системы почечных канальцев со многими лекарственными средствами обусловливает выведение как связанной, так и несвязанной их фракции из плазмы крови, а эффективность процесса удаления из крови большей части пропранолола печенью обеспечивается высокой степенью связывания препарата с белками плазмы.
Устойчивое состояние. При непрерывном введении лекарственного средства в условиях устойчивого состояния скорость введения будет равна скорости его выведения. Следовательно,
 при соответствующих размерностях единиц количества, объема и времени.
при соответствующих размерностях единиц количества, объема и времени.
Таким образом, если известен клиренс (С1), можно вычислить скорость введения, необходимую для достижения данного уровня содержания лекарственного средства в плазме крови. Определение клиренса лекарственного средства обсуждается в разделе, посвященном заболеваниям почек.
В том случае, если лекарственное средство вводят дробно, приведенную выше зависимость между его концентрацией в плазме крови и количеством, вводимым за один междозовый интервал, можно выразить следующим образом:

Средняя концентрация лекарственного средства в плазме крови (Срсредняя) отражает возможные колебания уровня содержания лекарственного средства в плазме крови (выше или ниже его среднего значения) во время междолевого интервала (см. рис. 64-2).
При пероральном введении лекарственного средства только некоторая доля (F) введенной дозы может попасть в систему кровообращения. Низкая биодоступность его может быть обусловлена неудачным изготовлением лекарственной формы, которая не распадается или не растворяется в жидкостях пищеварительного тракта. Существующие стандарты контроля за изготовлением лекарственных форм снизили остроту этой проблемы. Абсорбция лекарственных средств после перорального введения может подавляться взаимодействием различных препаратов. Биодоступность снижается также в результате метаболизма лекарственного средства в пищеварительном тракте и/или в печени во время процесса абсорбции, что носит название эффекта первичного происхождения и представляет особенно важную проблему для тех лекарственных веществ, которые обильно экстрагируются этими органами. Часто это приводит к значительной разнице в степени биодоступности таких лекарственных средств у разных больных. Лидокаин, применяемый для купирования аритмий, не назначают перорально именно вследствие наличия у него высокого эффекта первичного прохождения. Лекарственные средства, вводимые внутримышечно, также могут иметь низкую биодоступность (например, фенитоин). При возникновении-какой-либо неожиданной реакции на введение лекарственного средства следует рассмотреть в качестве возможной причины этого вопрос о его биодоступности. Это также следует учитывать при расчете дозового режима:

Выведение лекарственных средств, не следующих кинетике процессов первого порядка. Выведение некоторых лекарственных средств, таких как фенитоин, салицилаты и теофиллин, не следует кинетическим закономерностям процессов первого порядка в тех случаях, когда их количества в организме находятся в терапевтическом диапазоне. Клиренс подобных лекарственных препаратов изменяется по мере снижения уровней их содержания в организме во время процесса выведения или после изменений вводимой дозы. Такой процесс выведения называют дозозависимым. В соответствии с этим продолжительность времени, в течение которого концентрация лекарственного средства снижается наполовину, уменьшается по мере снижения уровня его содержания в плазме крови; это половинное время не является истинным периодом полувыведения, поскольку термин «период полувыведения» относится к кинетическим закономерностям процессов первого порядка и является величиной постоянной. Выведение фенитоина—дозозависимый процесс, и при очень высоких уровнях его содержания (в токсическом диапазоне) половинное время выведения может превышать 72 ч. По мере снижения концентрации препарата в плазме крови клиренс увеличивается и двукратное снижение концентрации его в плазме будет достигнуто через 20—30 ч. Если выведение лекарственного средства следует кинетическим закономерностям процессов первого порядка, между уровнем его содержания в плазме крови при устойчивом состоянии и величиной поддерживающей дозы существует прямая зависимость и удвоение дозы препарата должно привести к удвоению уровня его содержания в плазме крови. Однако если выведение лекарственных средств происходит в соответствии с кинетическими закономерностями дозозависимых процессов, увеличение вводимой дозы может сопровождаться непропорционально высоким возрастанием уровня содержания его в плазме крови. Так, при увеличении суточной дозы фенитоина с 300 до 400 мг уровень его содержания в плазме возрастает более чем на 33%. Степень этого увеличения непредсказуема, так как степень отклонения клиренса от закономерностей процесса первого порядка различна у разных больных. Выведение салицилатов при высоких уровнях их содержания в плазме крови также следует кинетическим закономерностям дозозависимого процесса, поэтому следует соблюдать осторожность при введении их в больших дозах, особенно детям. Метаболизм этанола также является дозозависимым процессом, что влечет за собой очевидные последствия. Механизмы, обусловлю вающие кинетические закономерности дозозависимых процессов, могут включать в себя насыщение, лимитирующее скорость метаболизма, или обратное угнетение продуктом реакции фермента, лимитирующего скорость метаболизма.
Дата добавления: 2015-07-25; просмотров: 58 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| ГЛАВА 63. ГЛАВНЫЙ КОМПЛЕКС ГЕНОВ ГИСТОСОВМЕСТИМОСТИ | | | Индивидуализация лекарственной терапии |