Читайте также:
|
2.1.Відбір і зберігання проб води
Відбір проби води є важливою частиною її аналізу, необхідною умовою для одержання правильних результатів і застосування їх на практиці. Помилки, які виникають внаслідок неправильного відбору проби, надалі виправити неможливо. Кількість проби, яку необхідно відібрати, залежить від числа компонентів, що визначають. Для детального аналізу необхідно відібрати мінімум 3 дм3 води.
Дня відбору проби найчастіше використовують скляні пляшки з прозорого безбарвного скла. Пляшки можуть бути циліндричної або призматичної форми з притертим скляним корком. Можна використати поліетиленові кришки. В окремих виняткових випадках можна використати гумовий корок, обгорнутий плівкою із полівінілхлориду або поліетилену.
Вибраний для проб посуд необхідно попередньо ретельно вимити. Для знежирення посуду застосовують синтетичні миючі речовини, потім його ретельно промивають водопровідною водою, ополіскують дистильованою і, нарешті, декілька разів водою, яку досліджують.
У більшості випадків пробу води можна відібрати в бутель. Якщо до води важко дістатися, бутель прикріпляють до держака або з додатковим тягарем опускають у воду на тросі.
Іноді необхідно взяти пробу води з певної глибини, не зміщуючи її з водою інших шарів. Для цього використовують спеціальні пристрої –батометри. Відбір середніх проб проточної води (із рік, потічків) здійснюють у місцях найсильнішої течії під поверхнею води на глибині 20–30 см.
Стоячі води водосховищ, озер і ставків неоднорідні за якістю у різних місцях і на різних глибинах. Тому пробу відбирають під поверхнею води на строго фіксованій глибині, уникаючи місць з густими заростями водяних рослин.
Із водопровідних кранів проби беруть так. На кран надягають гумовий шланг, другий кінець якого вводять в бутель для проби, опускаючи його до дна. Повільно відкривають кран, доки вода не потече безперервним струменем товщиною ~0,5 см. Після наповнення бутля водою його залишають ще деякий час під краном, щоб вода протікала через бутель до тих пір, поки температура її не стане постійною.
Із штучної водойми (ставка, озера) пробу беруть під поверхнею води безпосередньо у бутель.
При відборі проточної води з джерела пробу беруть безпосередньо у посудину, якщо у джерела немає водозбору та штучного стоку. Якщо джерело оснащене зливною трубою або жолобком, пробу відбирають у бутель з їх кінця.
При відборі проб з колодязя спочатку відкачують з нього воду до того часу, поки вода не буде мати постійну температуру (приблизно 20 хв.). Якщо колодязі не мають насосів, то пробу беруть без відкачування води із середньої частини водяного стовпа.
Слід уникати тривалого зберігання проб води.
Усі відібрані проби повинні мати супровідний документ (в нашому випадку у вигляді етикетки на бутлі).
Маркування проби води здійснюють за такою формою:
1. Проба № ________________________________________________________
2. Назва джерела (свердловина, джерело, колодязь, ріка тощо)______________
3. Адреса водопункту ________________________________________________
4. Глибина, на якій відібрана проба ____________________________________
5. Умови та методика відбору проби ___________________________________
6. Дата та час відбору проби __________________________________________
7. Прізвище, ініціали та посада особи, яка відбирала пробу ________________
2.2.Визначення аніонного складу домішок природних вод
2.2.1. Визначення вмісту іонів Cl-
Внаслідок значної розчинності у воді більшості хлоридів металів іони Cl- присутні майже в усіх водах. Вода, яка містить велику кількість хлоридів, має гіркувато-солоний смак і виявляє високу корозійну активність.
Метод визначення вмісту у воді хлоридів базується на осадженні іонів Cl- розчином аргентуму(I) нітрату AgNO3. Як індикатор використовують розчин калію хромату K2CrO4.
За присутності іонів Cl- спочатку під час додавання розчину AgNO3осаджується білий аргентуму(I) хлорид:
Cl- + AgNO3 = AgCl↓ + NO3- . (4)
Після зв’язування хлорид-іонів починає випадати осад аргентуму(I) хромату цегляно-червоного кольору:
K2CrO4 + 2 AgNO3 = Ag2CrO4↓ + 2 KNO3 . (5)
Кінець реакції зв'язування іонів Cl- фіксують за зміною забарвлення.
Послідовність визначення. У конічну колбу об’ємом 250 см3 відбирають піпеткою 20 см3 досліджуваної води, додають 1 см3 10%-го розчину K2CrO4, доводять об’єм розчину у колбі дистильованою водою до ~100 см3 і титрують 0,01N розчином AgNO3. Титрування ведуть на білому фоні, додаючи титрант AgNO3 порціями не більшими ніж 0,5 см3, а наприкінці титрування – краплями за постійного перемішування розчину, який титрують. Титрування закінчують після зміни кольору з зеленувато-жовтого на оранжево-цегляний. Виконують два паралельні титрування.
Вміст у воді хлорид-іонів  (мг/дм3) розраховують за формулою
(мг/дм3) розраховують за формулою
 (6)
(6)
де V1 – об'єм розчину AgNO3, витраченого на титрування проби води, см3;
V2 – об'єм проби води, см3;
 – нормальна концентрація розчину AgNO3, моль-екв/дм3;
– нормальна концентрація розчину AgNO3, моль-екв/дм3;
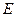 – еквівалентна маса Хлору (35,45 г/моль-екв).
– еквівалентна маса Хлору (35,45 г/моль-екв).
2.2.2. Визначення вмісту іонів HCO3-
Зазвичай гідрогенкарбонат-іони HCO3- є найпоширенішими аніонами природних вод.
Визначення вмісту гідрогенкарбонат-іонів базується на титруванні проби досліджуваної води хлоридною кислотою за присутності індикатора метил-оранжу. За наявності у воді гідрогенкарбонат-іонів відбувається реакція
HCO3- + HCl = Cl- + H2O + CO2 . (7)
Визначенню вмісту іонів HCO3- заважає наявність у воді іонів CO32- і ОН-,які також взаємодіють з хлоридною кислотою. Тому перед виконанням аналізу необхідно переконатися у відсутності цих аніонів у досліджуваній воді.
Послідовність визначення. У конічну колбу об’ємом 250 см3 відбирають піпеткою 100 см3 досліджуваної води, додають 2–3 краплі розчину індикатора фенолфталеїну. Якщо проба води залишається безбарвною (це свідчить про відсутність у воді іонів CO32- і ОН-), то у цю ж колбу додають 2–3 краплі розчину індикатора метилового оранжевого і титрують 0,1N розчином хлоридної кислоти до переходу забарвлення з жовтого на золотисто-рожеве.
Вміст у воді гідрогенкарбонат-іонів(мг/дм3) розраховують за формулою
 (8)
(8)
де V1 – об'єм розчину HCl, витраченого на титрування проби води, см3;
V2 – об'єм проби води, см3;
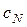 – нормальна концентрація розчину HCl, моль-екв/дм3;
– нормальна концентрація розчину HCl, моль-екв/дм3;
E – еквівалентна маса іонів HCO3- (60,98 г/моль-екв).
2.2.3. Визначення вмісту іонів SO42-
Наявність у природних водах сульфат-іонів зумовлена розчиненням осадових порід, окисненням гідрогену сульфіду H2S, а також забрудненням вод промисловими стоками. Підвищений вміст сульфатів у воді небажаний, оскільки сульфати металів порушують діяльність шлунково-кишкового тракту, така вода є корозійно активною.
Суть більшості кількісних методів визначення вмісту сульфат-іонів полягає в тому, що у досліджувану воду вводять іони Барію, які зв'язують сульфат-іони у вигляді важкорозчинного барію сульфату білого кольору
SO42- + Ba2+ = BaSO4↓ . (9)
Комплексонометричний метод визначення вмісту сульфат-іонів полягає у визначенні різниці витрати трилону Б (натрію етилендиамінацетату), який утворює комплексну сполуку з іонами Ba2+, до процесу осаджування сульфат-іонів і після їх осаджування. У зв'язку з тим, що у досліджуваній воді присутні іони Кальцію і Магнію, які також утворюють комплекси з трилоном Б, роблять відповідну поправку на ці іони.
Послідовність визначення. У конічну колбу об’ємом 250 см3 відбирають піпеткою 50 см3 досліджуваної води, доводять об’єм розчину у колбі дистильованою водою до ~100 см3 і додають 1–2 краплі індикатора метилового червоного. Далі краплями підкислюють розчин у колбі 0,1N розчином хлоридної кислоти до зміни забарвлення розчину на яскраво-червоне. Потім цей розчин кип'ятять 3–5 хв. До розчину, який кипить, додають піпеткою 2,0 см3 10%-го розчину барію хлориду, який містить іони Mg2+, і знову кип'ятять розчин протягом 10–15 с. Присутність у розчині барію хлориду іонів Mg2+ необхідна для чіткішого визначення кінця титрування. Частина іонів Ва2+ витрачається на зв'язування сульфат-іонів у малорозчинний барію сульфат, а частина залишається. Через 10–15 хв. досліджувану пробу води нейтралізують 0,1N розчином натрію гідроксиду, додаючи його обережно краплями до появи жовтого забарвлення. Потім додають ~5 см3 аміачного буферного розчину (суміш розчинів NH4Cl і NH4OH), 7–8 крапель індикатора хромогену чорного і титрують 0,1N розчином трилону Б (визначають об’єм V3)до появи синього (або зеленувато-синього) забарвлення розчину.
Окремо визначають об'єм трилону Б (V1), витраченого на титрування 2,0 см3 розчину барію хлориду. Для цього у конічну колбу об’ємом 250 см3 відбирають піпеткою 50 см3 дистильованої води, додають піпеткою 2,0 см3 10%-го розчину барію хлориду, ~5 см3 аміачного буферного розчину, 7–8 крапель індикатора хромогену чорного і титрують 0,1N розчином трилону Б до появи синього (або зеленувато-синього) забарвлення розчину.
Об'єм розчину трилону Б (V2), витраченого на титрування іонів Кальцію і Магнію у воді, приймають на основі визначення загальної твердості води з урахуванням взятого об'єму проби досліджуваної води (50 см3) (див. п. 2.3.1).
Вміст сульфат-іонів (мг/дм3) розраховують за формулою
 (10)
(10)
де V4 – об'єм проби води, см3;
 – нормальна концентрація розчину трилону Б, моль-екв/дм3;
– нормальна концентрація розчину трилону Б, моль-екв/дм3;
Е – еквівалентна маса сульфат-іона (48,03 г/моль-екв).
2.3.Визначення катіонного складу домішок природних вод
2.3.1. В изначення сумарного вмісту іонів Ca2+ і Mg2+
Наявність у природних водах іонів Кальцію і Магнію зумовлена розчиненням у воді мінералів, які містять іони Ca2+ і Mg2+, наприклад, гіпсу CaSO4 . 2H2O, а також розчиненням у водах, що містять СО2, таких природних мінералів: вапняку, мармуру, крейди (CaCO3), магнезиту MgCO3, доломіту CaCO3 . MgCO3 тощо за реакціями:
CaCO3 + H2O + CO2 D Ca(HCO3)2 ; (11)
MgCO3 + H2O + CO2 D Mg(HCO3)2 . (12)
Переважаючий вміст у природних водах іонів Кальцію, порівняно з іонами Магнію, пояснюють меншим вмістом сполук Магнію в осадових породах. Наявність у водах іонів Ca2+ і Mg2+ зумовлює їх твердість.
Найпоширенішим методом визначення сумарного вмісту у воді іонів Ca2+ і Mg2+ (загальної твердості води) є комплексонометричний. Суть цього методу полягає у зв’язуванні іонів Ca2+ і Mg2+ трилоном Б у складні комплексні сполуки в лужному середовищі. Утворення забарвлених комплексних сполук Кальцію і Магнію можливе лише за значень рН = 9–10, тому у досліджувану воду додають аміачно-буферний розчин. За вищих значень водневого показника (рН= 12–13) комплекси іонів Ca2+ з аніонами трилону Б є стійкими, а комплекси іонів Mg2+ в цьому середовищі руйнуються і Магній знаходиться у вигляді гідроксиду. Кінець титрування (точку еквівалентності) визначають за зміною кольору індикатора хромогену чорного, який з іонами Ca2+ і Mg2+ утворює комплекси винно-червоного кольору, а після зв’язування іонів Ca2+ і Mg2+ у комплекси з трилоном Б з’являється колір вільного індикатора – синій із зеленкуватим відтінком.
Послідовність визначення. У конічну колбу об’ємом 250 см3 відбирають піпеткою 50 см3 досліджуваної води, доводять об’єм розчину у колбі дистильованою водою до ~100 см3, додають ~5 см3 аміачно-буферного розчину, 6–7 крапель індикатора хромогену чорного і титрують 0,1N розчином трилону Б, інтенсивно перемішуючи розчин, до зміни забарвлення розчину з винно-червоного на синє із зеленкуватим відтінком. Виконують два паралельні титрування, за результатами яких визначають середнє значення об’єму розчину трилону Б, який витратили на титрування.
Сумарний вміст іонів Кальцію і Магнію  (ммоль-екв/дм3) розраховують за формулою
(ммоль-екв/дм3) розраховують за формулою
 (13)
(13)
де V1 – об'єм розчину трилону Б, витраченого на титрування, см3;
V2 – об'єм проби води, см3;
 – нормальна концентрація розчину трилону Б, моль-екв/дм3.
– нормальна концентрація розчину трилону Б, моль-екв/дм3.
2.3.2. Роздільне визначення вмісту іонів Ca2+ і Mg2+
Вміст іонів Кальцію визначають титруванням трилоном Б з індикатором мурексидом за рН= 12–13. Іони Кальцію з мурексидом утворюють комплекс рожевого кольору. Після зв’язування всіх іонів Кальцію в комплекс з трилоном Б з’являється колір вільного індикатора – бузково-фіолетовий. Після розкладу мурексиду кип’ятінням або бромною водою створюють середовище з рН = 9–10 і титрують іони Магнію трилоном Б з індикатором хромогеном чорним. Іони Магнію з індикатором хромогеном чорним утворюють комплекс бузково-фіолетового кольору. Після зв’язування всіх іонів Магнію в комплекс з трилоном Б з’являється колір вільного індикатора – синій із зеленкуватим відтінком.
Послідовність визначення. У конічну колбу об’ємом 250 см3 відбирають піпеткою 50 см3 досліджуваної води, доводять об’єм розчину у колбі дистильованою водою до ~100 см3, додають піпеткою 1,0 см3 10%-го розчину NaOH, ~15 мг суміші індикатора мурексиду і натрію хлориду (на кінчику скляної лопаточки). Розчин після перемішування титрують 0,1N розчином трилону Б до зміни забарвлення з рожевого на бузково-фіолетове. Відмічають об’єм розчину трилону Б (V1), який витрачено на титрування іонів Кальцію.
Потім до цієї ж колби додають 15 см3 0,1 N розчину хлоридної кислоти та кип’ятять пробу до зникнення забарвлення розчину. Після охолодження до проби додають ~5 см3 аміачно-буферного розчину і ~15 мг суміші індикатора мурексиду і натрію хлориду. Доводять об’єм розчину трилону Б у бюретці до нульової позначки і титрують іони Магнію розчином трилону Б до зміни забарвлення з бузково-фіолетового на синє. Відмічають об’єм розчину трилону Б (V2), який витрачено на титрування іонів Магнію. Виконують два паралельні титрування.
Вміст іонів Кальцію  (мг/дм3) розраховують за формулою
(мг/дм3) розраховують за формулою
 (14)
(14)
де V1 – об'єм розчину трилону Б, витраченого на титрування іонів Кальцію, см3;
V3 – об'єм проби води, см3;
– нормальна концентрація розчину трилону Б, моль-екв/дм3;
Е – еквівалентна маса Кальцію.
Вміст іонів Магнію  (мг/дм3) розраховують за формулою
(мг/дм3) розраховують за формулою
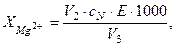 (15)
(15)
де V2 – об'єм розчину трилону Б, витраченого на титрування іонів Магнію, см3;
V3 – об'єм проби води, см3;
– нормальна концентрація розчину трилону Б, моль-екв/дм3;
Е – еквівалентна маса Магнію.
2.3.3. Визначення вмісту іонів Ca2+
Послідовність визначення. У конічну колбу об’ємом 250 см3 відбирають піпеткою 100 см3 досліджуваної води, додають піпеткою 2,0 см3 10%-го розчину NaOH, ~30 мг суміші індикатора мурексиду і натрію хлориду (на кінчику скляного шпателя) і титрують пробу розчином трилону Б до зміни забарвлення з рожевого на бузково-фіолетове. Відмічають об’єм розчину трилону Б (V1), який витрачено на титрування.
Вміст іонів Кальцію (мг/дм3) розраховують за формулою
 (16)
(16)
де V1 – об'єм розчину трилону Б, витраченого на титрування, см3;
V2 – об'єм проби води, см3;
 – нормальна концентрація розчину трилону Б, моль-екв/дм3;
– нормальна концентрація розчину трилону Б, моль-екв/дм3;
Е – еквівалентна маса Кальцію.
2.3.4. Визначення вмісту іонів Mg2+ розрахунковим шляхом
Вміст у воді іонів Мg2+ (ммоль-екв/дм3 і мг/дм3) розрахунковим шляхом визначають за різницею між сумарним вмістом іонів Кальцію і Магнію та вмістом іонів Кальцію:
 (17)
(17)
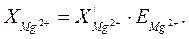 (18)
(18)
2.3.5. Визначення вмісту іонів Fe2+, Fe3+
Ферум завжди міститься у поверхневих і підземних водах у вигляді іонів Fe2+ і Fe3+. Підвищений вміст сполук Феруму у воді надає їй бурого забарвлення і неприємного присмаку. Вміст іонів Феруму у воді визначають колориметричним методом на основі реакції іонів Fe3+з калію або амонію роданідом. При цьому відбувається реакція
Fe3+ + 6 NH4SCN = [Fe(SCN)6]3- + 6 NH4+, (19)
у результаті якої утворюється яскраво-червоні комплексні іони [Fe(SCN)6]3-. Інтенсивність забарвлення проби води пропорційна концентрації іонів Fe3+.
За наявності у воді іонів Fe2+ їх попередньо окиснюють до іонів Fe3+ і визначають у такому стані. Інтенсивність забарвлення розчину вимірюють на фотоколориметрі.
Послідовність визначення. У конічну колбу об’ємом 250 см3 відбирають піпеткою 50 см3 досліджуваної води, додають піпеткою 1 см3 розведеної (1:1) хлоридної кислоти для створення кислого середовища (реакція (7.2) проходить повністю у кислому середовищі), 2–3 кристалики амонію персульфату для окиснення іонів Fe2+ до Fe3+. Нагрівають на водяній бані протягом 10 хв. (окиснення краще відбувається під час нагрівання), охолоджують колбу з розчином, потім додають 2 см3 50%-го розчину амонію або калію роданіду і вимірюють оптичну густину на фотоколориметрі (світлофільтр № 3, барабан вимірювання правий, довжина кювети 30 мм). Вміст іонів Феруму (мг/дм3) визначають за калібрувальним графіком.
Для побудови калібрувального графіка готують стандартний розчин, який містить 0,1 г/дм3 іонів Fe3+. Із цього розчину готують 7 проб (еталонні розчини) з концентраціями іонів Fe3+ відповідно 0,2; 0,4; 0,6; 0,8; 1,0; 1,2; 1,4 мг/дм3. У кожну пробу додають по 1,0 см3 хлоридної кислоти (1:1) і по 2,0 см3 розчину амонію або калію роданіду. Усі розчини забарвлюються у червоний колір різної інтенсивності.
На фотоколориметрі вимірюють оптичні густини еталонних розчинів (див. інструкцію роботи на фотоколориметрі). Будують калібрувальний графік, відкладаючи на осі абсцис концентрації іонів Fe3+, а на осі ординат – відповідні оптичні густини розчинів.
Вміст іонів Fe3+ у воді визначають аналогічно описаному вище визначенню сумарного вмісту іонів Fe2+ і Fe3+, але без додавання амонію або калію персульфату, тому розчин не нагрівають. Вміст у воді іонів Fe2+ визначають за різницею між сумарним вмістом іонів Fe2+ і Fe3+ і вмістом іонів Fe2+.
2.4.Розрахунки загальної мінералізації, сухого залишку та гіпотетичного сольового складу природних вод
2.4.1. Розрахунок загальної мінералізації та сухого залишку природних вод
Основна частина мінеральних солей, розчинених у природних водах, належить до сильних електролітів і повністю дисоціює на іони. Тому, за законом еквівалентів, сума ммоль-еквівалентів катіонів завжди дорівнює сумі ммоль-еквівалентів аніонів. Деякі розходження можливі лише за рахунок похибок виконання аналізів і неповноти визначення всіх іонів присутніх у воді.
На основі рівності суми ммоль-еквівалентів катіонів та аніонів розраховують сумарний вміст іонів Натрію і Калію, аналітичне визначення яких трудомістке.
Для розрахунку сумарного вмісту іонів Натрію і Калію  використовують експериментальні дані аналізів, виражені в ммоль-екв/дм3
використовують експериментальні дані аналізів, виражені в ммоль-екв/дм3
 (20)
(20)
У перерахунку на іони Натрію (мг/дм3) це становить
 (21)
(21)
Після цього складають таблицю катіонно-аніонного складу мінеральних домішок води (див. табл.1).
На основі одержаних даних катіонно-аніонного складу розраховують два показники: 1) загальну мінералізацію (загальний вміст солей); 2) сухий залишок.
Загальну мінералізацію  визначають за сумою вмісту катіонів і аніонів, її виражають в мг/ дм3
визначають за сумою вмісту катіонів і аніонів, її виражають в мг/ дм3

 (22)
(22)
На основі одержаного значення загальної мінералізації роблять висновок про те, до якої категорії вод належить досліджувана вода (прісна, солонувата, солона тощо). Прісні води, придатні для господарсько-питного і технічного водопостачання, характеризуються загальною мінералізацією до 1000 мг/дм3.
Сухий залишок можна також розрахувати за даними катіонно-аніонного складу досліджуваної води. Оскільки під час випаровування проби води за температури І05°С кальцію і магнію гідрогенкарбонати розкладаються за реакцією
Ca(HCO3)2 = CaCO3↓ + H2O↑ + CO2↑, (23)
або в молекулярно-іонній формі
2 HCO3– = CO32– + H2O↑ + CO2↑ (24)
2 . 61 г/моль 60 г/моль
то в сухий залишок переходять лише карбонат-іони (М= 60 г/моль), тобто лише приблизно половина від вмісту гідрогенкарбонат-іонів. Тому під час розрахунку сухого залишку вміст гідрогенкарбонат-іонів множать на ~0,5 (точніше на 30/61).
Сухий залишок  (мг/дм3) розраховують за формулою
(мг/дм3) розраховують за формулою
 (25)
(25)
2.4.2. Приклад розрахунку загальної мінералізації
Дата добавления: 2015-07-12; просмотров: 51 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| ТЕОРЕТИЧНИЙ ВСТУП | | | та сухого залишку природних вод |