Читайте также:
|
Помимо гипогликемии, у больных диабетом часто наблюдают и два других острых метаболических осложнения — диабетический кетоацидоз и гиперосмолярную некетозную кому. Первое —- это осложнение инсулинзависимого диабета, а второе встречается обычно
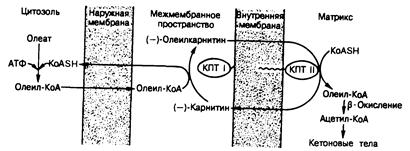
Рис. 327-4. Система карнитинпальмитоилтрансферазы, транспортирующая длинноцепочечные жирные кислоты в митохондрии.

Рис. 327-5. Регуляция кетогенеза.
Образование в печени больших количеств ацетоацетата и бета-гидроксибутирата требует достаточного притока свободных жирных кислот (в качестве субстрата) и активации их окисления. Причиной липолиза служит в основном дефицит инсулина, тогда как путь окисления жирных кислот активируется главным образом глюкагоном. Непосредственной причиной ускоренного окисления служит падение содержания манония-КоА. (По J. D. McGarry, D. W. Foster, Amer. J. Med., 61:9, 1976)
при инсулиннезависимом диабете. При истинном диабете II типа кетоацидоз, если и встречается, то чрезвычайно редко.
Диабетический кетоацидоз. Диабетический кетоацидоз возникает, по-видимому при инсулиновой недостаточности и относительном или абсолютном повышении концентрации глюкагона. Это осложнение часто проявляется при отмене инсулина, но может индуцироваться также физическим (например, инфекцией, хирургической операцией) или психическим стрессом даже на фоне продолжающейся инсулинотерапии. В первом случае при отмене инсулина повышается концентрация глюкагона, тогда как при стрессе провоцирующим фактором служит, вероятно, адреналин и/или норадреналин. Выброс адреналина не только стимулирует секрецию глюкагона, но и, предположительно, блокирует остаточную секрецию небольших количеств инсулина, сохраняющуюся у некоторых больных с
ИЗСД, и тем самым ингибирует вызываемое инсулином поглощение глюкозы периферическими тканями. Эти гормональные сдвиги вызывают множество нарушений в организме, но два из них особенно неблагоприятны: это 1) максимальная стимуляция глюконеогенеза и нарушение периферической утилизации глюкозы и 2) активация процесса кетогенеза.
1. Максимальная стимуляция глюконеогенеза и нарушение периферической утилизации глюкозы приводит к выраженной гипергликемии. Глюкагон облегчает глюконеогенез, вызывая снижение уровня фруктозо-2,6-дифосфата—интермедиата, который стимулирует гликолиз за счет активации фосфофруктокиназы и блокирует глюконеогенез вследствие ингибирования фруктозодифосфатазы. При снижении концентрции фруктозо-2,6-дифосфата гликолиз тормозится, а глюконеогенез усиливается. Возникающая гипергликемия вызывает осмотический диурез, что приводит к уменьшению объема жидкости и дегидратации, столь характерным для кетоацидоза.
2. Активация процесса кетогенеза и тем самым индукция метаболического ацидоза. Чтобы возник кетоз, изменения должны затрагивать как жировую ткань, так и печень. Основным субстратом образования кетоновых тел служат свободные жирные кислоты из жировых запасов. Если кетогенез ускоряется, то повышается концентрация свободных жирных кислот в плазме. Однако если печеночные механизмы окисления жирных кислот не активированы, то жирные кислоты, поступающие в печень, реэстерифицируются и либо запасаются в форме печеночных триглицеридов, либо превращаются в липопротеины очень низкой плотности и вновь попадают в кровоток. Хотя высвобождение жирных кислот усиливается из-за недостатка инсулина, более быстрое окисление их в печени обусловлено в основном глюкагоном, влияющим на систему карнитинацилтрансферазы (фермента, обеспечивающего транспорт жирных кислот в митохондрии после их эстерификации коэнзимом А). Как показано на рис. 327-4, карнитинацилтрансфераза I (карнитинпальмитоилтрансфераза I) трансэстерифицирует жирный ацилКоА в жирный ацилкарнитин, который уже свободно проникает через внутреннюю мембрану митохондрий. Обратная реакция происходит внутри митохондрий и катализируется карнитинацилтрансферазой II (карнитинпальмитоилтрансферазой II). У сытого человека карнитинацилтрансфераза I неактивна, в результате чего длинноцепочечные жирные кислоты не могут вступить в контакт с ферментами b-окисления, что необходимо для образования кетоновых тел. При голодании или некомпенсированном диабете система активна; в этих условиях скорость кетогенеза оказывается функцией первого порядка концентрации жирных кислот, достигающих трансферазы I.
Глюкагон (или изменение соотношения глюкагон/инсулин) активирует систему транспорта двумя путями. Во-первых, он вызывает быстрое падение уровня малонил-КоА в печени. Этот эффект обусловлен блокадой последовательности реакций глюкозо-6-фосфат ® пируват ® цитрат ® ацетил-КоА ® малонил-КоА вследствие упомянутого выше снижения уровня фруктозо-2,6-дифосфата. Малонил-КоА — первый важный интермедиат на пути синтеза жирных кислот из глюкозы, является конкурентным ингибитором карнитинацилтрансферазы 1, и снижение его концентрации активирует этот фермент. Во-вторых, глюкагон вызывает повышение концентрации карнитина в печени, что по закону действующих масс сдвигает реакцию в сторону образования жирного ацилкарнитина. Указанные процессы схематически суммированы на рис. 327-5. При высокой концентрации жирных кислот в плазме их захват печенью оказывается достаточным, чтобы насытить как окислительный, так и эстерифицирующий пути, что приводит к ожирению печени, гипертриглицеридемии и кетоацидозу. Главной причиной кетоза служит чрезмерное образование кетонов в печени, но определенную роль играет и ограничение периферической утилизации ацетоацетата и b-гидроксибутирата.
Клинически кетоз проявляется потерей аппетита, тошнотой, рвотой и повышением скорости образования мочи. Могут возникать боли в животе. Без надлежащего лечения могут иметь место нарушение сознания и кома. При обследовании обращают на себя внимание дыхание Куссмауля и признаки уменьшения объема жидкости в организме. Последнее редко достигает степени, достаточной для развития коллапса сосудов и прекращения функции почек. При неосложненном кетоацидозе температура тела остается нормальной или снижается, лихорадка же указывает на наличие инфекции. Лейкоцитоз, часто очень выраженный, характерен для диабетического ацидоза как такового и необязательно свидетельствует об инфекции.
Характерные для диабетической комы метаболические нарушения перечислены в табл. 327-10. Некоторые из них целесообразно прокомментировать. Метаболический ацидоз и избыток анионов почти всегда обусловлен повышенным уровнем ацетоацетата и b-гидроксибутирата в плазме, хотя определенное значение имеют и другие кислоты (например,
Дата добавления: 2015-07-25; просмотров: 69 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| II. Ежедневная терапия | | | Т а блица 327-10. Начальные лабораторные признаки диабетического кетоацидоза |