Читайте также:
|
Курт Дж. Иссельбахер (Kurt J. Isselbacher)
Определение. Термином «галактоземия» обозначают два вида врожденных нарушений обмена галактозы: классическая галактоземия обусловливается недостаточностью галактозо-1-фосфатуридилтрансферазы (ГАЛТ) и сопровождается катарактой, умственной отсталостью и циррозом печени, а недостаточность галактокиназы обусловливает главным образом образование катаракты.
Патогенез. Лактоза — основной углевод молока — представляет собой дисахарид, содержащий галактозу и глюкозу. В пищеварительных путях она гидролизуется кишечной лактазой. В норме всосавшаяся галактоза в печени превращается в глюкозу. Первой реакцией на этом пути является фосфорилирование галактозы с образованием галактозо-1-фосфата под действием галактокиназы, кодируемой геном, расположенным на 17-й хромосоме:

На следующем этапе галактозо-1-фосфат превращается в глюкозо-1-фосфат под действием ГАЛТ, ген которой расположен на 9-й хромосоме:
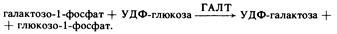
УДФ-сахара могут превращаться один в другой в процессе эпимеразной реакции:
УДФ-галактоза Û УДФ-глюкоза.
Галактоза метаболизируется и другими путями. В присутствии НАДФ•Н (или НАД-Н) она может превращаться (восстанавливаться) в галактитол (дульцитол) под действием альдозоредуктазы. Небольшое ее количество может и окисляться галактозодегидрогеназой с образованием галактоновой кислоты, ксиулозы и двуокиси углерода. Эти пути и определяют ограниченный метаболизм галактозы у больных с галактоземией.
При недостаточности галактокиназы галактоза накапливается в крови и тканях. В хрусталике глаза она под действием альдозоредуктазы превращается в галактитол — сахар, для которого хрусталик непроницаем. В результате происходит чрезмерная гидратация, которая на фоне уменьшения количества глутатиона в хрусталике обусловливает развитие катаракты.
При классической галактоземии недостаточность ГАЛТ приводит к накоплению в тканях галактозо-1-фосфата и галактозы. Как и при недостаточности галактокиназы, катаракта развивается вследствие накопления в хрусталике галактитола. Предполагают, что цирроз печени и задержка психического развития связаны с увеличением количества галактозо-1-фосфата в этих тканях. Повышенный уровень галактозы в крови может снижать печеночную продукцию глюкозы, обусловливая гипогликемию. В почках и кишечнике накопление галактозы и галактозо-1 -фосфата приводит, по-видимому, к торможению транспорта аминокислот. У некоторых женщин нарушается функция яичников в связи с гипергонадотропным гипогонадизмом, патогенез которого неизвестен.
Как недостаточность галактокиназы, так и недостаточность ГАЛТ наследуются по аутосомному рецессивному типу. У гетерозигот по этим нарушениям уровни ферментов составляют лишь половину от нормы, но симптоматика болезни отсутствует. Недостаточность галактокиназы у беременной, потребляющей лактозу, может обусловить развитие катаракты у плода. Однако не все больные с недостаточностью ГАЛТ в клетках бывают носителями классической галактоземии. У некоторых лиц, гомозиготных по другому гену, называемому вариантом Дуарте, уровень ГАЛТ обычно также в 2 раза ниже нормы, но симптоматика при этом отсутствует. Их можно отличить от больных с классической галактоземией по данным исследования электрофоретических свойств мутантного фермента. Как при недостаточности галактокиназы, так и при классической галактоземии может быть выявлена либо функциональная недостаточность, либо отсутствие необходимого фермента. Классическая галактоземия обусловливается мутацией структурного гена, поэтому измененный фермент (ГАЛТ) лишен нормальной функции. Известны и другие клинические варианты с измененной электрофоретической подвижностью фермента.
Частота классической галактоземии среди представителей европеоидной популяции составляет примерно 1:80000 новорожденных. Около 0,8—1,3 % населения представляют собой гетерозиготы по гену галактоземии (ГАЛТ) и примерно 10 % — носители варианта Дуарте. При скрининге новорожденных на галактоземию наиболее частой причиной патологии служит компаунд-гетерозиготность по варианту Дуарте и классической галактоземии, при которой содержание ГАЛТ находится на уровне 17 % от нормы. Клинические проявления у них отсутствуют.
Клинические проявления. Симптомы классической галактоземии обычно появляются в первые несколько дней или недель после рождения. Ребенок отказывается от грудного молока или молочных продуктов, у него появляются рвота и признаки недоедания, его развитие замедляется. Могут присоединиться желтуха, гепатомегалия и признаки печеночной патологии. Обычно у новорожденных катаракта не выявляется, но развивается через несколько недель или месяцев. Отсталость психического развития становится очевидной в возрасте 6—12 мес. У детей с классической галактоземией часто развивается бактериальный сепсис (особенно обусловленный кишечной палочкой), который может быть основной причиной смерти новорожденного. Единственным постоянным симптомом недостаточности галактокиназы служит катаракта.
Диагностика. Недостаточность галактокиназы следует подозревать у младенцев или детей с катарактой, в моче которых определяют отличающиеся от глюкозы восстанавливающие вещества. Диагноз устанавливают при выявлении дефицита галактокиназы в эритроцитах.
О классической галактоземии следует думать, если налицо один из упомянутых признаков или более. При потреблении молока в моче больного появляется восстанавливающий сахар, неопределяемый с помощью глюкозоксидазной реакции (т. е. не глюкоза), но идентифицируемый как галактоза с помощью других (например, хроматографических) методов. Если у ребенка появляется рвота, снижается аппетит или ему вводят глюкозу внутривенно, то галактоза в моче может и не определяться. Окончательный диагноз устанавливают, убедившись в недостаточности или отсутствии ГАЛТ в эритроцитах. Для этого используют разные методы. Заболевание можно диагностировать и пренатально, исследуя ферменты в культуре клеток, полученных при амниоцентезе, или обнаружив увеличенное количество галактитола в амниотической жидкости.
В неонатальном периоде галактоземию следует дифференцировать от первичной печеночной патологии. При болезни печени нарушается извлечение галактозы из крови и может повышаться уровень галактозы в крови и в моче. Однако в этих случаях содержание ГАЛТ остается в пределах нормы.
Лечение. Оно заключается в исключении из диеты продуктов, содержащих галактозу, особенно молока. Часто используют заменители молока, такие как нутрамиген. Несмотря на то что препараты из бобов сои содержат галактозу в составе полисахаридов, они, по-видимому, хорошо переносятся больными, так как связанная галактоза высвобождается очень медленно. У больных детей, вскармливаемых препаратами сои, уровень галактозо-1-фосфата в эритроцитах, как правило, не повышается.
Перевод больного на безгалактозную диету обычно сопровождается резким ослаблением симптоматики, за исключением задержки психического развития. Больного следует содержать на безгалактозной диете в течение неопределенно продолжительного периода или, по крайней мере, до тех пор, пока не нормализуется их физическое и неврологическое состояние.
Другие нарушения углеводного обмена. Признаки наследственного нарушения толерантности к фруктозе и недостаточности фруктозо-1,6-дифосфатазы — двух видов аутосомных рецессивных нарушений метаболизма фруктозы, приводящих к гипогликемии, суммированы в табл. 314-1 (см. также гл. 313 и 329).
Таблица 314-1. Некоторые другие нарушения углеводного обмена
| Нарушение | Метаболический дефект | Проявления |
| Наследственное нарушение толерантности к фруктозе | Недостаточность фруктозо-1-фосфатальдолазы приводит к накоплению фруктозо-1-РО4 в тканях | Печеночная патология, повреждение почечных канальцев и гипогликемия |
| Недостаточность фруктозо-1,6-дифосфатазы | Недостаточность фермента блокирует глюконеогенез из обычных предшественников — лактата, глицерина и аланина, поэтому уровень глюкозы в крови зависит от поступления экзогенной глюкозы | Лактацидоз приводит к гипервентиляции, сонливости и коме обычно на фоне гипогликемии и кетоза |
Дата добавления: 2015-07-25; просмотров: 68 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| Болезни с индивидуальной патофизиологией | | | ГЛАВА 315. ГИПЕРЛИПОПРОТЕИНЕМИИ И ДРУГИЕ НАРУШЕНИЯ ЛИПИДНОГО ОБМЕНА |