Читайте также:
|
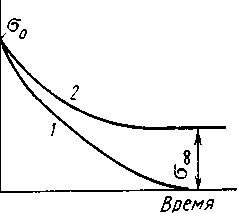
Рисунок 2.1 – Релаксация напряжения в линейном (1) и пространственно сшитом (2) эластомере
Через определенный промежуток времени происходит перегруппировка всех напряженных узлов флуктуационной сетки, что дает возможность
клубкам макромолекул перейти в прежнее свернутое статистически наиболее вероятное состояние. В этот момент напряжение в образце падает до нуля, а структура образца становится такой же, как и до растяжения. Но сама деформация растяжения не изменилась – она по-прежнему равна 100%. Это может произойти только в том случае, если в процессе сворачивания клубков и перегруппировки узлов флуктуационной сетки клубки одновременно смещались относительно друг друга. Такой процесс есть процесс течения макромолекул. Теперь после освобождения-образца из зажимов динамометра он более не сократится Эластическая деформация в 100%, которой был подвергнут образец вначале вся перешла в деформацию течения.
Чем более полярен полимер, тем более пологой оказывается кривая релаксации — медленнее падает напряжение.
Если эластомер пространственно сшит (вулканизован), т. е. на ряду
с флуктуационной сеткой в нем образовалась сетка химических связей то эта последняя практически не релаксирует. Правильнее сказать, что она релаксирует много медленнее, чем флуктационная сетка. В пространственно-сшитом образце напряжения релаксируют до тех пор, пока все они не сосредоточатся на узлах химической сетки. При этом все узлы флуктационной сетки оказываются разгруженными. Напряжение в образце достигает предела (кривая 2 рис. 2.1). Химические связи препятствуют необратимому перемещению клубков молекул, но не препятствуют перемещению сегментов. Если образец освободить из зажимов динамометра, то он с течением времени полностью восстановит свои первоначальные размеры и только тогда напряжение в нем упадет до нуля.
В опыте по релаксации напряжения в растянутом образце, как мы видели, эластическая обратимая деформация со временем переходит в вязкоте- кучую необратимую. Полностью обратимая деформация развивается в идеально упругой стальной пружине, а полностью необратимая деформация развивается при нагружении поршня, помещенного в идеальную жидкость. По
следовательное соединение пружины и поршня является простейшей моделью вязкоупругого тела (рис. 2.2). Эта модель носит название модели Максвелла (по имени ее создателя).
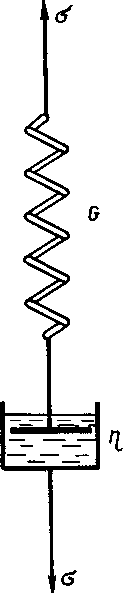
Рисунок 2.2 - Модель Максвелла
Процесс релаксации напряжения описывается уравнением Максвелла:
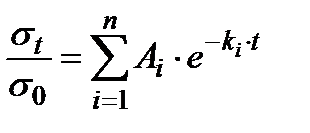
где σt и σ0 – напряжение растяжения в образце в момент времени t и начальный момент соответственно;
Ai – коэффициент, учитывающий долю i-го процесса релаксации;
ki – константа скорости i-го процесса релаксации.
2.2 Ползучесть
Для изучения релаксационных явлений образец быстро нагружают и следят за ходом деформации растяжения под действием приложенной нагрузки. При этом сложность состоит в том, что поперечное сечение образца со временем уменьшается и таким образом одна и та же нагрузка вызывает в расчете на постоянно уменьшающуюся площадь поперечного сечения все
возрастающее напряжение. Существует немало остроумных способов уменьшения действующей силы (груза) по мере растяжения образца, так чтобы сохранить неизменным во времени действующее напряжение. В конечном счете ползучесть осуществляется в режиме σ=const, ε =f(t). На рисунке 2.3 показана типичная кривая ползучести линейного полимера (кривая 1).
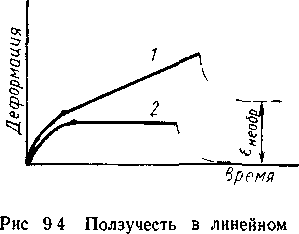
Рисунок 2.3 – Ползучесть в линейном (1) и пространственном полимере.
Под действием нагрузки макромолекулярные клубки развертываются, часть сегментов перемещается, ориентируясь в направлении действия силы. Перемещение сегментов приводит также к смещению клубков относительно друг друга. Таким образом, с самого момента нагружения образца в нем развиваются одновременно и обратимая высокоэластическая деформация, и деформация необратимая вязкотекучая.
Если после некоторого времени образец разгрузить, то он частично сократится за счет свертывания клубков макромолекул. Это и будет величина высокоэластической де формации, развившейся к моменту разгрузки
Однако полного сокращения не происходит, сохраняется некоторая остаточная деформация, являющаяся истинно необратимой вязко текучей.
Под действием заданного напряжения клубки макромолекул развернутся тем больше, чем меньше их упругость. Возникшая высокоэластическая деформация далее остается неизменной. Перемещением клубков относительно друг друга течение не прекратится вязкотекучая деформация будет нарастать во времени с постоянной скоростью. Поэтому кривая ползучести делится на два участка начальный криволинейный, когда развиваются оба типа деформации и линейный участок когда накапливается только вязкотекучая деформация. Строго говоря, наличие линейного участка характерно только для полимера с одним временем релаксации. Для реального полимера можно наблюдать участок кривой кажущийся линейным за время наблюдения, но в действтельностни обусловленный различными типами молекулярных перемещений с очень большими временами релаксации.
На том же рис. 2.3приведена кривая ползучести идеального сетчатого эластомера (кривая 2) в нем не возникает необратимая деформация из за наличия прочных химических связей исключающих взаимное перемещение макромолекул. Эластическая деформация осуществляется лишь в той мере в какой позволяет сетка химических связей ползучесть развивается, достигая предела. После разгрузки образец сокращается до первоначальных размеров.
Основная особенность кривой ползучести — наличие участка замедленного развития упругой деформации. В реальном полимере упругая деформация развивается не мгновенно как в пружине, а замедленно так как перемещение сегментов тормозится вязким сопротивлением среды.
Указанная особенность — наличие замедленной упругости (высокоэластичности) — отображается моделью Кельвина — Фойхта рис. 2.4
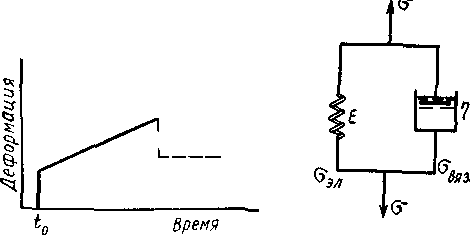
Рисунок 2.4 – Модель Кельвина - Фойхта
Здесь пружина и поршень соединены параллельно напряжения на них складываются 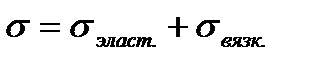 . По закону Гука
. По закону Гука 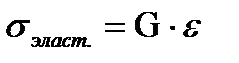 , по закону вязкого
, по закону вязкого
течения Ньютона 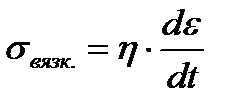 . В результате
. В результате 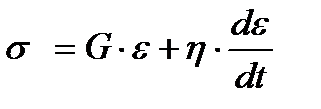 , откуда после интегрирования получаем
, откуда после интегрирования получаем 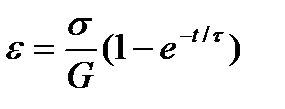 . Это уравнение не предусматривает наличия необратимой деформации поэтому лучше всего моделирует ползучесть пространственно сшитого эластомера. Ползучесть линейного полимера хорошо описывается также объединенной механической моделью сочетающей модель Максвелла и модель Кельвина - Фойхта.
. Это уравнение не предусматривает наличия необратимой деформации поэтому лучше всего моделирует ползучесть пространственно сшитого эластомера. Ползучесть линейного полимера хорошо описывается также объединенной механической моделью сочетающей модель Максвелла и модель Кельвина - Фойхта.
2.3 Релаксационные процессы в полимерах
В процессах релаксации участвуют различные структурные элементы полимера, каждый из которых имеет свои времена релаксации, лежащие в широком интервале — от 10-12 с до многих лет. Релаксационные свойства полимеров зависят от типа и размера релаксирующих в данных условиях элементов структуры. Поэтому для полной характеристики полимеров необходимо рассматривать сумму всех релаксационных процессов, образующих так называемый релаксационный спектр. Эти спектры получают или статическими методами (релаксация напряжения) или динамическими (механические или диэлектрические потери).
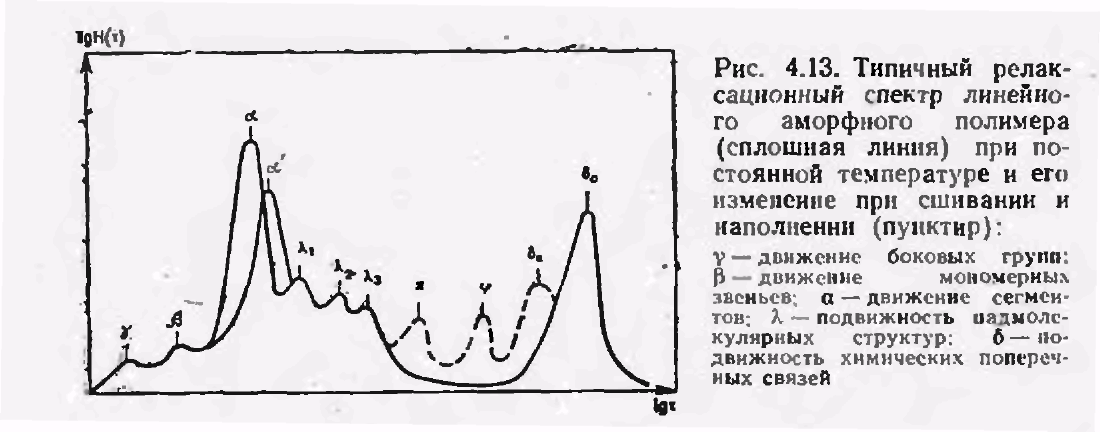
Рисунок 2.5 – Типичный релаксационный спектр линейного аморфного полимера (сплошная линия) при постоянной температуре и его изменение при сшивании и наполнении (пунктир): γ – движение боковых групп; β – движение мономерных звеньев; α – движение сегментов; λ – подвижность надмолекулярных структур; δ – подвижность химических поперечных связей
На рисунке 2.5 приведен типичный спектр времен релаксации линейного полимера. Максимумы на релаксационном спектре соответствуют временам релаксации τi* каждой релаксируюшей единицы при постоянной температуре. Каждое дискретное время, характеризующее данный переход, зависит от температуры и снижается при ее повышении вследствие увеличения подвижности элементов структуры:
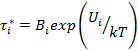
где Вi — константа, зависящая от размеров структурного элемента; Ui — энергия активации i-й структурной единицы, равная высоте энергетического барьера, преодолеваемого при переходе структурного элемента из одного равновесного состояния в другое.
В общем случае процесс релаксации напряжения при заданной деформации описывается уравнением
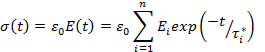
где σ(t) и E(t)—напряжения и модуль через время t при деформации ε0; τi* — время релаксации i-й кинетической единицы; n — число кинетических единиц различных типов; Ei — коэффициент, соответствующий вкладу i-ой кинетической единицы в общий процесс релаксации.
Среднее значение времен релаксации всех структурных единиц называется средним временем релаксации τср*.
Неоднородность структуры полимера, введение наполнителей и других компонентов, соединение макромолекул друг с другом, например при образовании пространственной сетки в процессе вулканизации каучуков, приводит к увеличению числа структурных элементов, влияющих на релаксационные процессы. В макромолекуле, содержащей разные по структуре и размерам боковые группы, вместо одного максимума γ-процесса имеется несколько (γ1, γ2 и т д) с низкой энергией активации (10—20 кДж/моль). Для гомополимера характерен один β-переход, а для блок-сополимера уже два — β1 и β2. В кристаллических полимерах одна и та же структурная единица обладает различной подвижностью в аморфной и кристаллической фазах, соответственно наблюдаются два перехода β и β1 характерные для аморфной и кристаллической структуры. Например, в спектре полиэтилена высокой плотности проявляются β- и β1-переходы при 150 и 250 К и частоте 1000 Гц. Очень часто вместо одного а- и λ-максимумов появляется несколько, что обусловлено различной подвижностью сегментов (а-переход) или надмолекулярных структур (λ-переход). Например, в кристаллических полимерах (полиэтилене) существует даже три максимума - а, а1 и а2, обусловленные сегментальной подвижностью в аморфной фазе, переходных аморфно-кристаллических межфазных слоях 'и аморфных участках лучей сферолитов.
λ-Переходы, обусловленные подвижностью надмолекулярных структур, относятся к медленным релаксационным процессам и связаны с наличием физической сетки. В некоторых полимерах могут существовать физические узлы различной природы, например в бутадиен-нитрильных эластомерах проявляется так называемый π-процесс релаксации, обусловленный диполь-дипольным взаимодействием групп 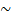 CN---NC~ (при 363 К).
CN---NC~ (при 363 К).
При образовании водородных связей в спектре обнаруживается переход, аналогичный π-переходу, но при более высокой температуре. Переходы, обусловленные релаксацией химических связей между молекулами (δ), также могут проявляться в виде нескольких максимумов, если существуют связи различной энергии. Так, в серных резинах наряду с переходом δс-с имеет место переход δs, обусловленный подвижностью (т. е. разрывом) химических серных связей.
В наполненных полимерах вместо одного а-перехода появляется еще один — а', обусловленный подвижностью сегментов макромолекул, адсорбированных поверхностью наполнителя, а также φ-переход, который связывают с подвижностью частиц наполнителя в среде полимера. В таблице 2.1 приведены характеристики релаксационных процессов в резинах на основе наполненного сополимера бутадиена со стиролом.
Таблица 2.1- Характеристики релаксационных процессов в резине на основе сополимера бутадиена и стирола (70:30) с 20% технического углерода П-234
| Релаксационный процесс | Характерное время релаксации при 293К, τi,с | Энергия активации Ui,кДж/моль | Константа Bi, с | Объем кинетической единицы Vi, см3 | Линейные размеры кинетической единицы, нм |
| β | 1*10-8 | 5*10-13 | 2*10-22 | 0,5 | |
| α | 6*10-6 | 5*10-12 | 1*10-21 | 3-4 | |
| α’ | 1,5 | 4,8*10-12 | 1*10-21 | 3-4 | |
| λ | 1,6*102 | 2,6*10-8 | 2,0*10-17 | ||
| 2,1*103 | 3,4*10-6 | 4*10-16 | |||
| 4,3*104 | 6,9*10-6 | 1,6*10-14 | |||
| φ | 2*105 | 1,6*10-8 | 0,9*10-17 | ||
| δ | 4*109 | 2*10-13 | ≈10-23 | 0,2-0,3 |
Вклад каждого релаксационного процесса зависит от его характерам условий проявления. Деформация полимера в стеклообразном состоянии при Т<ТС сопровождается релаксационными β- и γ-переходами. Энергия активации этих переходов намного меньше, и поэтому высота их пиков намного ниже, чем в случае α-перехода. Они оказывают существенное влияние на механические свойства полимеров в стеклообразном состоянии. При растяжении в высокоэластнческом состоянии наибольшее влияние на релаксацию оказывают не γ- и β-, α α-, α'-, λ-, δ- и φ-переходы, т. е. процессы ориентации сегментов (τ=10-6 — 10-3с), перегруппировки надмолекулярных структур (τ=102— 104 с), перегруппировки поперечных химических связей и связей в цепях (τ=107—109с) и др.
Однако влияние этих факторов на скорость релаксационных процессов зависит от продолжительности действия силы. Как уже упоминалось, вероятность проявления гибкости макромолекулой зависит от соотношения времени действия силы и времени, необходимого для изменения конформации. Последнее есть не что иное, как время релаксации τ*, поэтому можно считать, что реакция полимера на механическое воздействие определяется соотношением между временем релаксации и временем деформации τ*/t (рисунок 2.7). Если τ*/t 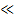 1, то система очень быстро релаксирует и приходит в равновесное состояние. Это условие может быть реализовано или при очень малых значениях τ*, или при очень высоких значениях t. Для полимеров этот случай имеет место при высоких температурах (τ* снижается) или очень низких скоростях воздействия. При τ*/t
1, то система очень быстро релаксирует и приходит в равновесное состояние. Это условие может быть реализовано или при очень малых значениях τ*, или при очень высоких значениях t. Для полимеров этот случай имеет место при высоких температурах (τ* снижается) или очень низких скоростях воздействия. При τ*/t 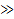 1 релаксация протекает очень медленно, что может быть следствием или высоких значений τ* (система малоподвижна, например в стеклообразном или кристаллическом состояниях), или высоких скоростей механического воздействия. При τ*/t→1 релаксационные процессы развиваются за какой-то определенный промежуток времени и оказывают наибольшее влияние на свойства материала. Таким образом, релаксационные явления проявляются для всех рассмотренных физических состояний — стеклообразного, вязкотекучего и высокоэластического, но в наибольшей степени релаксация характерна для высокоэластического состояния.
1 релаксация протекает очень медленно, что может быть следствием или высоких значений τ* (система малоподвижна, например в стеклообразном или кристаллическом состояниях), или высоких скоростей механического воздействия. При τ*/t→1 релаксационные процессы развиваются за какой-то определенный промежуток времени и оказывают наибольшее влияние на свойства материала. Таким образом, релаксационные явления проявляются для всех рассмотренных физических состояний — стеклообразного, вязкотекучего и высокоэластического, но в наибольшей степени релаксация характерна для высокоэластического состояния.
Если сопоставить влияние температуры и времени действия силы, то можно отметить, что отношение τ*/t снижается либо при повышении температуры (τ* уменьшается), либо при увеличении t, т. е. существует эквивалентность влияния времени и температуры. Этот принцип получил название принципа температурно-временной суперпозиции (ТВС). Любая релаксационная характеристика при изменений температуры от Т до Т’ изменяется на величину K, равную
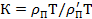
(ρп и ρп' — плотность полимера при Т и Т').
На основании принципа ТВС, зная температурные зависимости какого-либо показателя, отражающего релаксационные свойства в узком интервале скоростей воздействия, можно предсказать, как будет изменяться это свойство при любой заданной температуре при изменении скорости воздействия. На рисунке 2.7 показано использование принципа ТВС для гипотетического полимера при релаксации напряжения. Кривая при определенной температуре, например при 273 К, выбирается в качестве отсчетной; эта температура называется приведенной Тпр. Затем все кривые сдвигаются вдоль логарифмической шкалы времени до их наложения. Кривые при Т>Тпр сдвигаются вправо, а кривые при Т<Тпр влево. Обобщенная кривая при Тпр охватывает значительно больший период времени, чем экспериментальные данные. Расстояние, на которое сдвигались соседние кривые вдоль оси абсцисс, называется фактором приведения аТ.
Температурная зависимость фактора приведения аТ описывается уравнением Вильямса — Ландела — Ферри (ВЛФ):
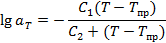
(С1 и С2—эмпирические константы).
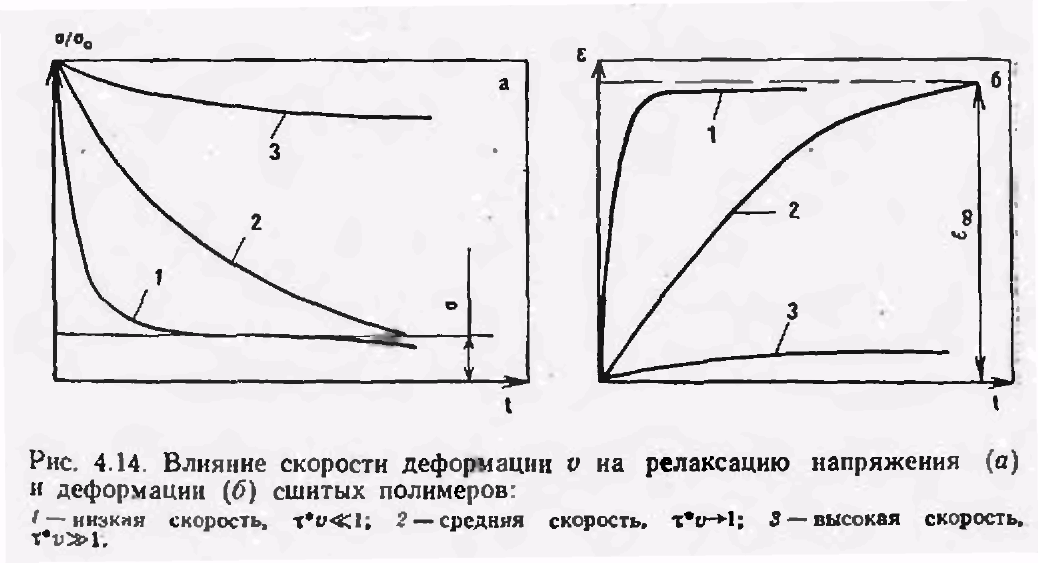
Рисунок 2.6 – Влияние скорости деформации υ на релаксацию напряжения (а) и деформацию (б) сшитых полимеров: 1 – низкая скорость τ*υ  1; 2 – средняя скорость τ*υ→1; 3 – высокая скорость τ*υ
1; 2 – средняя скорость τ*υ→1; 3 – высокая скорость τ*υ 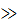 1
1
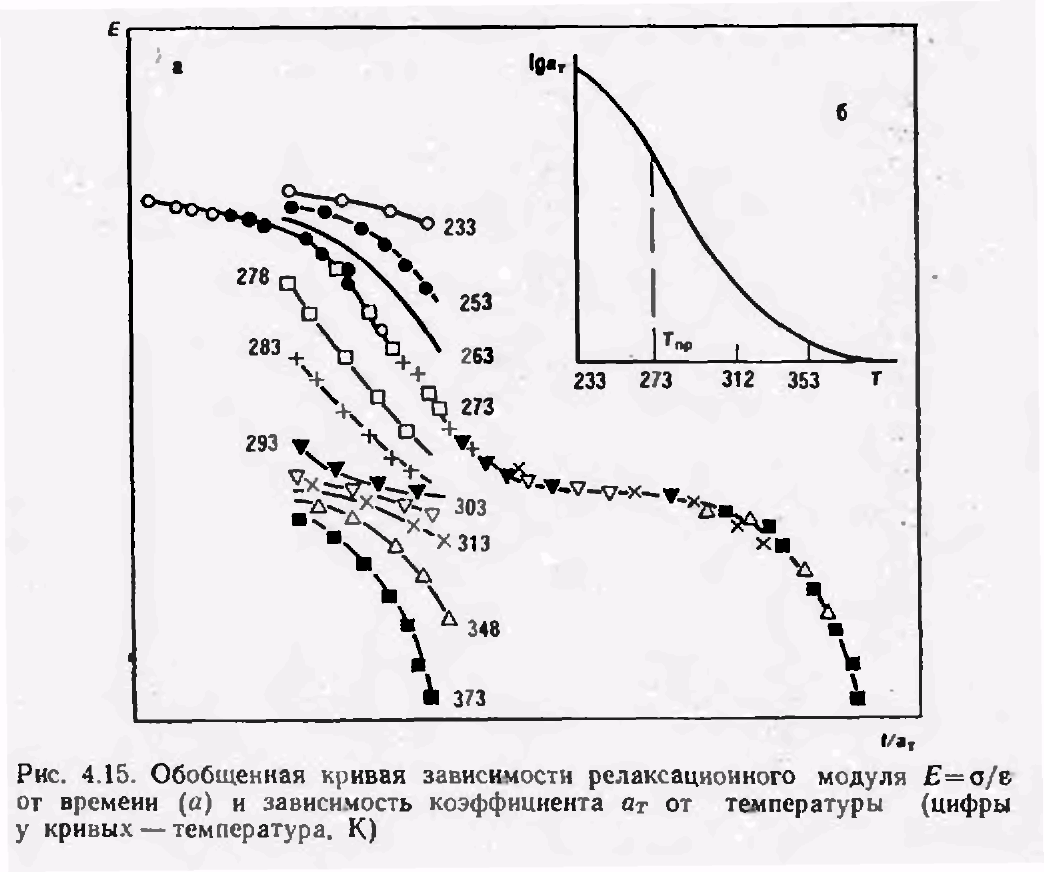
Рисунок 2.7 – Обобщенная кривая зависимости релаксационного модуля Е=σ/ε от времени (а) и зависимость коэффициента аТ от температуры (цифры у кривых – температура, К)
В качестве температуры приведения Тпр часто выбирают Тс. В этом случае для большинства аморфных полимеров аТ примерно одинаково и равно
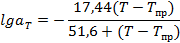
Если же за Тпр выбирается другая температура, то константы С1 и С2 будут иметь другие значения.
2.4 Различные типы релаксационных переходов в полимерах
Релаксационные явления в значительной степени определяют протекание физических и химических процессов в полимерах. Полимеры — сложные многоуровневые системы, состоящие из структурных элементов (кинетических единиц) различной природы (атомов, боковых и концевых групп, звеньев макромолекул, свободных и связанных сегментов, элементов надсегментальной и надмолекулярной структуры, физических и химических узлов сетки, частиц наполнителя и т. д.). Это приводит к большому разнообразию форм молекулярной подвижности и соответствующих им релаксационных процессов, которые наблюдаются при действии на полимер механических, электрических или магнитных полей. При этом наиболее универсальным воздействием, позволяющим получить полную информацию о молекулярной подвижности и процессах релаксации в полимерах, является механическое воздействие. Электрические и магнитные поля могут вызвать не все релаксационные переходы, так как электрическое поле действует только на элементы, обладающие дипольным моментом, а магнитное поле — на элементы, обладающие магнитным моментом.
Релаксационные процессы (переходы) связаны с различными формами теплового движения в полимере, характеризуемого спектром молекулярной подвижности структурных элементов. Этому спектру соответствуют
кретные τ1, τ2,… τn и непрерывные Н(τ) спектры времен релаксации (рисунок 2.9). Дискретному спектру τi соответствует п релаксационных переходов, проявляющихся в виде максимумов на непрерывном спектре Н(τ). Максимумы на спектре внутреннего трения χ(Т) соответствуют дискретному спектру температур Тi релаксационных переходов (i=1, 2,..., n) (рисунок 2.10).
Из температурных зависимостей τi и частотных зависимостей Тi можно получить сведения о размерах кинетических единиц, участвующих в каждом релаксационном процессе, и особенностях их внутреннего строения, а также сведения об энергиях активации процессов, зависящих от характера взаимодействия кинетических единиц и прочности их сцепления.
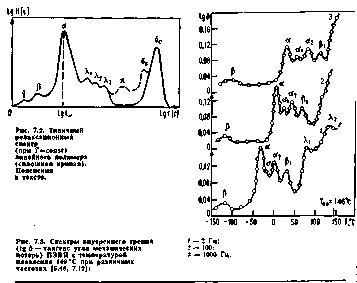
Рисунок 2.8 – Типичный релаксационный спектр
Рисунок 2.9 – Спектр внутреннего трения
В связи с этим релаксационная спектрометрия выступает как структурный метод, позволяющий выявить все возможные релаксационные переходы, характерные для данного полимерного материала, и выяснить их природу, т. е. природу структурных элементов, участвующих в переходе. Поскольку
Релаксационным процессам соответствуют максимумы как на спектрах внутреннего трения χ(Т) или χ(ω), так и на спектрах времен релаксации Н(τ). Эти спектры могут быть получены как квазистатическими методами (релаксация напряжения), так и динамическими методами (механические потерн, диэлектрические потери и т. д.).
Максимумы на релаксационных спектрах (см. рисунок 2.8) соответствуют временам релаксации дискретного спектра τi при заданной температуре Т=const, а максимумы на спектрах внутреннего трения (см. рисунок 2.9) соответствуют дискретным температурам релаксационных переходов Ti при заданной частоте деформации ω = const или дискретным частотам переходов ωi при заданной температуре T=const. По частотным зависимостям температур Ti и температурным зависимостям частот ωi могут быть рассчитаны времена релаксации τi. В целом дискретный спектр времен релаксации характеризуется временами T1, T2,..., Тn и вкладами E1,E2,…En отдельных релаксационных процессов. При этом доля каждого вклада не зависит от температуры.
Каждое дискретное время, характеризующее данный релаксационный переход, зависит от температуры по известному уравнению Больцмана — Аррениуса:
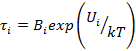
где Ui — энергия активации i-того процесса, зависящая от сил взаимодействия кинетических единиц, а k—коэффициент, зависящий от их размеров.
Энергии активации различны для разных релаксационных переходов, но практически для всех наблюдаемых релаксационных процессов не зависят от температуры в пределах точности измерений, за исключением α-процесса (стеклование).
Экспериментальные данные по дискретному релаксационному спектру могут быть представлены в координатах lg τi-1/T в виде семейства прямых, как это следует из уравнения. Тогда из наклонов прямых находятся Ui, а из отрезков, отсекаемых на оси 1/T=0,— коэффициент Bi. Независимым методом эти постоянные можно определить из данных спектра внутреннего трения (динамический метод) по зависимости температуры перехода Ti от частоты деформации ω = 2πν, где ν — линейная частота. Так, температуре перехода соответствует известное условие:
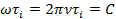
где С – безразмерная константа, С≈1 для мелкомасштабных движений (атомы, группы атомов в γ-, β-, δ-процессах) и С≈10 для сегментов и более крупных единиц.
Учитывая первое уравнение, получим:
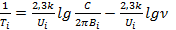
Отложив экспериментальные данные в координатах 1/Ti—Igν, находят значения Ui и Bi, а затем вычисляют τi. Только в том случае, когда оба метода дают совпадающие результаты по числу релаксационных переходов n и по величинам τi, полученный дискретный спектр релаксационных переходов считается надежным и отражающим структурные особенности полимера.
Релаксационным переходам в полимерах посвящено много обстоятельных исследований, результаты которых суммированы в монографиях. Работы последних лет привели к четкой классификации релаксационных переходов в аморфных и кристаллических полимерах. Все релаксационные переходы можно разбить на несколько групп, каждая из которых характеризуется молекулярной подвижностью одного и того же структурного элемента.
К первой группе относятся мелкомасштабные релаксационные процессы, связанные с подвижностью боковых привесков в макромолекулах (γ-группа). Потеря подвижности (колебательно-вращательных движений) этих кинетических единиц происходит при низких температурах (обычно ниже 200 К). Энергия активации мала (10—20 кДж/моль), а коэффициент Вi в уравнении примерно равен 10-13 с. Разные по структуре и размерам боковые группы в линейных полимерах отвечают γ1-, γ2- релаксационным переходам и т.д. Эти переходы обнаруживаются по механическим и диэлектрическим потерям при высоких частотах, так как времена релаксации τi для этих процессов малы (10-9—10-10 с при 300 К), а частоты велики (с=1 и ν≈10e Гц). По терминологии, принятой для диэлектрической релаксации в полимерах, процессы этой группы называются дипольно-групповыми γ-процессами.
Ко второй группе относятся мелкомасштабные процессы, связанные с подвижностью малых участков полимерной цепи — звеньев (β-группа). Процессы этой группы протекают в аморфной фазе полимера, находящегося в стеклообразном состоянии, когда сегментальная подвижность еще заморожена. На рисунке 2.9 виден β-переход для полиэтилена (Tβ= - 146°С) при ν=l Гц. С точки зрения реакции на действие электрических переменных полей (диэлектрическая релаксация), β-процесс— это локальное движение полярных групп, связанное с изгибными колебаниями малых групп в основной цепи (дипольно-групповые β-процессы). Для полиэтилена Вi= 1,6- 10-13 с, Ui = 30 кДж/моль и процесс связывается с подвижностью групп СН2 в цепи полиэтилена. Так как Bi~vi — объему кинетической единицы, то для ПЭ ui≈(2-4)10-20 мм3. Для разных полимеров Bi различаются мало, но энергии активации могут принимать значения до 80 кДж/моль.
Таким образом, процессы γ- и β-групп относятся к мелкомасштабным релаксационным процессам. Если полимерная цепь имеет боковые группы одного типа, то наблюдается один γ-процесс, если двух типов, то два γ-процесса. Для аморфного гомополимера наблюдается один β-переход, а для блок-сополимера— два перехода. В кристаллических полимерах одна н та же
К третьей группе относятся α-переходы, кинетической единицей которых является сегмент с Bi =5*10-12 с для гибкоцепных полимеров. Это значение Bi соответствует объему сегмента Vi≈10-13 мм3. В аморфных полимерах наблюдается один α-переход (см. рисунок 2.9), ответственный за стеклование полимера; если полимер наполнен активным наполнителем, то появляется еще один— α’-процесс с тем же значением Bi, но с энергией активации большей, чем для α-процесса. Например, для изопренового эластомера СКИ-3 при 300 К Ua =33 кДж/моль, a Ua’ = 62-67 кДж/моль в зависимости от типа наполнителя; для бутадиен-стирольного эластомера СКС-30 АРКМ-15 при 300К Ua = 34 кДж/моль, a Ua’=71 кДж/моль.
В кристаллическом полимере (полиэтилене) обнаруживаются кроме а-перехода еше два перехода (см. рисунок 2.9) с тем же значением Вi = 5-10-12 с. Это указывает на то, что α1- и α2-переходы также связаны с сегментальной подвижностью, но сегмент находится в других структурных условиях. Основной «-переход ответственен за стеклование аморфной фазы полимера. Для ПЭ Ua=51,5 кДж/моль и характеризует сегментальную подвижность в аморфной фазе полимера. Переходы α1 и α2 с Uai =54,5 кДж/моль и Uα2=59 кДж/моль связаны с сегментальной подвижностью в переходных аморфно-кристаллических межфазных слоях и в аморфных участках лучей сферолитов. В кристаллической фазе сегментальная подвижность не проявляется (Uα=  ). Для некристаллических гибкоцепных полимеров (эластомеров, пластмасс) характерные значения Uа=30 - 50 кДж/моль, а времена релаксации τa = = 10-6 - 10-3 с при З00К.
). Для некристаллических гибкоцепных полимеров (эластомеров, пластмасс) характерные значения Uа=30 - 50 кДж/моль, а времена релаксации τa = = 10-6 - 10-3 с при З00К.
В переменных электрических полях наблюдаются аналогичные механическим диэлектрические дипольно-сегментальные потери, природа которых та же—сегментальная подвижность. В полимерных стеклах сегментальная подвижность играет важную роль, так как является причиной многих явлений (стеклование, вынужденная высокоэластичность, ползучесть, квазихрупкое разрушение, трещины «серебра» и т. д.). В кристаллических полимерах сегменты могут находиться в трех различных состояниях, а в наполненном аморфном полимере — в двух состояниях, что приводит к мультиплетности релаксационных спектров a-процесса релаксации. Основным при этом остается a-процесс, ответственный за стеклование. Его вклад, как можно судить по высоте максимумов на спектрах, существенно больше, чем остальных процессов этой группы.
К четвертой группе относятся λ-процессы релаксации, наблюдаемые
выше Тс в зоне высокоэластического плато. Наблюдается как минимум три процесса: λ1, λ2, λ3, но в сополимерах их может быть больше. Эти переходы обычно выражены слабо по сравнению с a-переходом и характеризуются тем, что коэффициенты Вi относительно велики (10-8— 10-5 с) —превышают Вi = 5*10-12 с для сегментов на 3—6 порядков. При этом λi - процессы имеют одну и ту же энергию активации, совпадающую с энергией активации вязкого течения полимера, но различные Вi. Большие значения Вi указывают на то, что эти процессы связаны с подвижностью крупных структурных элементов, характерных для надсегментальных и надмолекулярных микроструктур, имеющих вид упорядоченных микрообластей (структурных объемных микроблоков, кластеров).
В связи с этим структуру аморфных полимеров (полимерных стекол и эластомеров) можно представить состоящей из двух структурных частей: неупорядоченной части, включающей свободные сегменты и цепи (их подвижность проявляется в α-процессах), и упорядоченной части, распространенной по всему объему в виде упорядоченных микрообластей флуктуационного происхождения (структурных микроблоков глобулярного, складчатого и мицеллярного типов различных линейных размеров — от 10 до 100 нм).
Структурные микроблоки играют роль физических узлов молекулярной сетки, имеющих различные, относительно большие времена жизни, которые соответствуют дискретным временам λi процессов релаксации (τi= 102 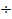 10+4 с при 300 К и Uλ =
10+4 с при 300 К и Uλ = 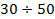 кДж/моль для различных линейных полимеров и объемом λ-микрообластей 10-14-10-15 мм3). Относительно низкие для структурных микроблоков, включающих тысячи и десятки тысяч «связанных» сегментов, значения Uλ, объясняются тем, что распад и образование таких упорядоченных микрообластей происходит посегментально путем отрыва и прилипания отдельных сегментов, аналогично испарению и конденсации капли жидкости, происходящим путем отрыва и прилипания отдельных молекул.
кДж/моль для различных линейных полимеров и объемом λ-микрообластей 10-14-10-15 мм3). Относительно низкие для структурных микроблоков, включающих тысячи и десятки тысяч «связанных» сегментов, значения Uλ, объясняются тем, что распад и образование таких упорядоченных микрообластей происходит посегментально путем отрыва и прилипания отдельных сегментов, аналогично испарению и конденсации капли жидкости, происходящим путем отрыва и прилипания отдельных молекул.
Процессы λ-релаксации ответственны за медленные релаксационные процессы в высокоэластическом состоянии, к которым обычно относят ползучесть, медленную стадию релаксации; напряжения и вязкое течение, а процесс α-релаксации в этой же температурной области ответственен за быстрые релаксационные процессы. Во всех этих процессах, а также в процессе вытяжки и ориентации, основную роль играет релаксация физических узлов молекулярной сетки.
λ-процессы релаксации ответственны за механизм разрушения эластомеров. В полярных полимерах природа физических узлов молекулярной сетки может быть иной. Например, в бутадиен-нитрильных эластомерах между λ- и δ-процессами обнаруживается π-процесс релаксации (см. рисунок 2.8), связанный с распадом и рекомбинацией локальных физических поперечных связей, обусловленных диполь-дипольным взаимодействием между полимерными цепями CN…NC. Температура перехода этого процесса Тл=90°С, энергия активации 90 кДж/моль, Вi= 10~10 с.
Пятая группа релаксационных переходов, следовательно, связана с подвижностью диполь-дипольных поперечных связей в полимерах.
Как следует из предыдущих глав, в твердых полимерах разрушение во многих случаях является следствием термофлуктуационного разрыва химических связей. Возникает вопрос, как связаны между собой процессы разрушения и релаксационные процессы в полимерах. Обратимся к химическому процессу релаксации (δ-процесс). На релаксационном спектре (см. рисунок 2.9) представлен δc-процесс — процесс химической релаксации, связанный с распадом связей С—С. Сразу отметим, что указанный релаксационный спектр получен из изотерм релаксации напряжения при малых деформациях и напряжениях, далеких от разрывных. Поэтому энергия активации Uс должна быть близка к энергии активации процесса разрушения Uo и энергии термодеструкции ED.
Дискретный спектр сшитого бутадиенметилстирольного эластомера СКМС-30 (рисунок 2.10) показывает, что вклад в релаксацию напряжения б-процесса очень велик по сравнению с вкладом λ-процессов, число которых больше трех благодаря микроблочному строению цепей сополимеров.
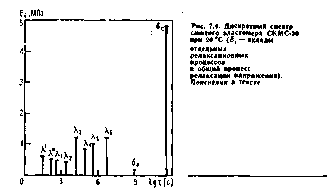
Рисунок 2.10 – Дискретный спектр
Для этого эластомера энергия активации U0c = 143 кДж/моль, а коэффициент Bi=3,2*10-4с. Столь малое значение Вi, меньшее, чем для γ- и δ-процессов, свидетельствует о том, что этот процесс является мелкомасштабным. Период колебаний атомов углерода в связях С—С, по данным ИКС, τ0≈ 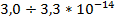 с, что совпадает с значением Bi. А так как в формуле Bi имеет смысл периода колебания, то тем самым доказано, что кинетической единицей в δс-процессе является атом углерода в полимерной цепи.
с, что совпадает с значением Bi. А так как в формуле Bi имеет смысл периода колебания, то тем самым доказано, что кинетической единицей в δс-процессе является атом углерода в полимерной цепи.
По данным масс-спектрометрического термического анализа (МТА) для СКМС-30, энергия активации термодеструкции связей С—С на первой стадии 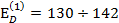 кДж/моль, что близко к энергии активации δс-процесса релаксации. Данные МТА для ПММА:
кДж/моль, что близко к энергии активации δс-процесса релаксации. Данные МТА для ПММА: 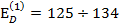 кДж/моль, а энергия активации разрушения U0=134 кДж/моль. Итак, можно сказать, что энергии активации трех процессов: химической релаксации, термической деструкции и процесса разрушения (при ϭ→0) совпадают.
кДж/моль, а энергия активации разрушения U0=134 кДж/моль. Итак, можно сказать, что энергии активации трех процессов: химической релаксации, термической деструкции и процесса разрушения (при ϭ→0) совпадают.
Для эластомера СКМС-30, сшитого серой, возникает дополнительный процесс химической релаксации δs (см. рисунок 2.9) с небольшим вкладом (см. рисунок 2.11). Коэффициент Bi = = 6,6*10-14 с, а энергия активации Uδs = 126 кДж/моль. Суть этого процесса, состоит в непрерывной перестройке сетки путем разрыва и рекомбинации
К шестой группе релаксационных переходов, таким образом, относятся химические процессы релаксации.
Дата добавления: 2015-08-20; просмотров: 172 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| V. ВИСНОВОК | | | ИССЛЕДОВАНИЕ ПРОЦЕССА ОТБОРА КАТОДНОГО ТОКА |