мыши таким образом, что оно приобретает некоторые сегменты антитела человека, сохраняя в то же время свою исходную антигенсвязывающую специфичность. Такое гибридное антитело называют химерным. Первым участком моноклонального антитела мыши, который был заменен соответствующим участком антитела человека, был Fc-фрагмент. Выбор объяснялся тем, что Fc-фрагмент антитела мыши выполнял роль эффектора иммунного ответа у человека недостаточно хорошо; кроме того, он с большой вероятностью индуцировал образование антител в организме человека. Чтобы снизить иммуногенность и усилить эффекторные функции, провели замену последовательностей ДНК, кодирующих Fv-области L- и Η-цепей иммуноглобулина человека, на аналогичные фрагменты специфического моноклонального антитела мыши (рис. 10.9). Такую замену можно осуществить разными путями: реплицировать ДНК in vitro с применением олигонуклеотидов в качестве затравки либо использовать субклониро-ванные фрагменты ДНК. Сегменты ДНК, кодирующие химерные цепи, встраивали в экспрессирующий вектор и вводили в культуру В-лимфоцитов, из которой выделяли наработанные антитела.
Химерные антитела, несущие антигенсвязы-вающий участок моноклонального антитела мыши к поверхностному антигену клеток рака толстой кишки человека, тестировали на больных с раком толстой и прямой кишки. Антитела оставались в кровотоке примерно в шесть раз дольше обычных антител мыши, тем самым оказывая свое действие в течение большего времени. При этом лишь у одного пациента из 10 наблюдался слабо выраженный иммунный ответ. К сожалению, в этих испытаниях не удалось получить противоопухолевого эффекта антител; возможно, это было связано с введением их в слишком малых дозах или с тем, что раковый процесс находился на поздних стадиях. В опытах in vitro химерные антитела проявляли высокую эффекторную активность, что позволяет надеяться на успешное их применение в других случаях.
Конструирование химерных молекул, о которых шла речь выше, — это первый этап в создании моноклональных антител мышей и крыс, обладающих сходством с антителами человека. Другой подход состоит в замещении только CDR-участков человеческих антител фрагмента-

| Рис. 10.9. Полученное методом генной инженерии антитело, сходное по своей структуре с антителом человека. Участки генов L- и Η-цепей иммуноглобулина человека, кодирующие VL- и vh -домены, заменены последовательностями ДНК, кодирующими VL- и VH-домены иммуноглобулина мыши. Продуктом рекомбинантных генов является химерный иммуноглобулин, обладающий антигенсвязывающей специфичностью моноклонального антитела мыши, эффекторными свойствами Fc-фрагмента иммуноглобулина человека и пониженной иммуногенностью для человека. |
Микробиологическоепроизводство лекарственных средств 217
| Рис. 10.10. Полученное методом генной инженерии антитело, сходное по своей структуре с антителом человека. Сегменты ДНК, кодирующие CDR-участки (CDR1, CDR2 и CDR3) антитела человека, заменены на последовательности, кодирующие CDR-участки Н- и L-цепей иммуноглобулина мыши. Продуктом этого рекомбинантного гена является иммуноглобулин с антигене вязы вающей специфичностью моноклонапьного антитела мыши (участки, выделенные розовым цветом) и остальными свойствами антитела человека (участки, выделенные голубым цветом). | 
|
ми моноклональных антител грызунов (рис. 10.10). Такие «восстановившие форму» антитела человека могут стать эффективным терапевтическим средством, поскольку они по своей антигенсвязывающей способности приближены к исходным моноклональным антителам грызунов.
Моноклональные антитела грызунов, сходные с антителами человека, можно получить, выделив кДНК L- и H-цепей из клеточной линии гиб p ид омы грызунов и амплифицировав их вариабельные области с помощью ΠЦР. В качестве праймеров для амплификации можно использовать олигонуклеотиды, комплементарные высококонсервативным сегментам ДНК, фланкирующим с 5'- и 3’-концов последовательность, кодирующую вариабельную область. Зная нуклеотидные последовательности кДНК вариабельных областей легкой и тяжелой цепей (VL и VH), легко определить границы CDR, основываясь на том, что соответствующие им последовательности гипервариабельны, в то время как каркасные области относительно консервативны. Исходя из данных о нуклеотидных последовательностях ДНК, кодирующих CDR грызунов, синтезировали шесть пар олигонуклеотидных праймеров. Каждая пара инициировала синтез ДНК, кодирующей одну из шести CDR грызунов: три, локализованных на L-цепи, и три — на Η-цепи. Кроме того, на 5'-конце каждого прай-мера находилось 12 дополнительных нуклеотидов, комплементарных фланкирующим последовательностям каркасных участков ДНК человека, по которым происходило встраивание CDR-ДНК грызунов (рис. 10.11). Далее с помощью олигонуклеотид-направленного мутагенеза осуществляли последовательную замену CDR-ДНК человека амплифицированной CDR-ДНК грызунов — фактическую «пересадку» CDR от грызунов в каркасные участки молекулы антитела человека. Модифицированную таким образом кДНК антител встраивали в векторы экспрессии и трансформировали ими подходящие клетки-хозяева, обычно Е. colt или клетки млекопитающих, в которых и вырабатывались антитела.
Данный метод предполагает, что за антигенсвязывающую способность антитела отвечают только CDR-участки, а не каркасные области. Однако, если связывание «гибридного» антитела с антигеном происходит недостаточно эффективно, может возникнуть необходимость в замене некоторых аминокислот в каркасных областях с помощью олигонуклеотид-направленного мутагенеза.
К настоящему времени этим методом получено более 50 различных моноклональных антител, обладающих сходством с антителами человека. К сожалению, данная технология, являясь весьма эффективной и универсальной, довольно дорогостоящая и требует больших затрат вре-
218 ГЛАВА 10

|
| Рис. 10.11. ПЦР-амплификация CDRl-участка, входящего в состав кДНК L-цепи моноклонального антитела грызуна. Олигонуклеотидные праймеры Р1 и Р2 комплементарны последовательностям ДНК CDRl-участка антитела грызуна. Кроме того, каждый праймер содержит на 5'-конце 12 нуклеотидов, комплементарных каркасным участкам (framework region, FR) кДНК L-цепи антитела человека. Используя шесть пар олигонуклеотидных праймеров — три для VL- и три для Vн-областей, — с помощью ПЦР амплифицировали отдельно каждый из ДНК-сегментов, кодирующих CDR-участки антитела грызуна. Затем, используя олигонуклеотид-направленный мутагенез, заменили ими соответствующие сегменты генов антитела человека. Такая замена оказалась возможной благодаря тому, что амплифицированные фрагменты содержали участки, комплементарные каркасным областям гена антитела человека. |
мени. Возможно, более предпочтительным способом получения антител человека и их фрагментов окажется метод, основанный на использовании фаговых «комбинаторных» библиотек, созданных на основе мРНК, полученной из В-клеток неиммунизированных доноров.
Производство антител с помощью Е. соli
Гибридомы, подобно большинству других клеточных культур животных, растут относительно медленно, не достигают высокой плотности и требуют сложных и дорогих сред. Получаемые таким образом моноклональные антитела очень дороги, что не позволяет широко использовать их в клинике. Чтобы решить эту проблему, были предприняты попытки создания своего рода «биореакторов" на основе генетически модифицированных бактерий, растений и животных. Для эффективной доставки и функционирования некоторых иммунотерапевтических средств зачастую достаточно одной антигенсвязывающей области антитела (Fab- или Fv-фрагмента), т. е. присутствие Fc-фрагмента антитела необязательно.
На рис. 10.12 представлена методика получения функциональных антител с помощью E. coli (рис. 10.12).
1. Используя мРНК, выделенную из вырабатывающих антитела клеток (В-лимфоцитов) мыши или человека, синтезируют кДНК.
2. Проводят раздельную ПЦР-амплифицикацию кДНК, кодирующих Н- и L-цепи.
3. Амплифицированные кДНК обрабатывают специфическими рестрицирующими эндонуклеазами, а затем встраивают в вектор на основе бактериофага λ. кДНК Н- и L-цепей содержат разные, характерные для каждой из них эндонуклеазные сайты, что облегчает специфическое встраивание каждой нуклеотидной последовательности в свой вектор. На этом этапе происходит клонирование множества разных сегментов Н- и L-цепей (рис. 10.13, А и Е).
4. кДНК одной Н- и одной L-цепи встраивают в общий «комбинаторный» вектор, так что в бактериофаге синтезируются обе цепи и образуется «полноценный» Fv-фрагмент (рис. 10.13,5).
Синтез Н- и L-цепей происходит во время литического цикла бактериофага λ, поэтому можно провести скрининг библиотеки клонов комбинаторных бактериофагов с целью определения их антигенсвязывающей активности.
Микробиологическое производство лекарственных средств 219

|
| Рис. 10.12. Создание с помощью Е. соli комбинаторной библиотеки кДНК VL- и Vн-областей антитела. |
На этапе соединения кДНК Н- и L-цепей в одном векторе образуется широкий спектр генов различных антител. Некоторые из них кодируют уникальные сайты связывания, получить которые с помощью обычной гибридомной технологии было бы невозможно. Пул антител млекопитающих включает 106—108 разных антител. Фаговая библиотека содержит примерно столько же клонов, поэтому можно ожидать, что одна комбинаторная библиотека будет вырабатывать такое же количество различных антител (Fv-молекул), как любое млекопитающее. Кроме того, однажды создав исходную комбинаторную библиотеку, можно комбинировать L- и Η-цепи и получать Fv-фрагменты, распознающие необычные эпитопы. Еще большего разнообразия можно достичь, используя неспецифический мутагенез. Поскольку за относительно короткое время можно провести скрининг миллионов фаговых бляшек, идентификация Fv-фрагментов с нужной специфичностью занимает от 7 до 14 дней. Для сравнения: скрининг нескольких сотен гибридомных клеточных линий обычно занимает месяцы.
Векторы на основе бактериофага λ не очень пригодны для получения больших количеств белковых молекул. Чтобы решить эту проблему, сконструировали такой вектор, в котором ДНК Н-и L-цепей встраиваются в сайт, фланкированный плазмидной ДНК. Такую плазмиду, содержащую ДНК Н- и L-цепи, можно вырезать из вектора и трансформировать ею E. coli (рис. 10.12). Являясь частью плазмиды, ДНК Fv-фрагментов будет многократно реплицироваться в клетках Е. coli cобразованием большого количества продукта, который можно использовать как в диагностических, таки в терапевтических целях.
При создании комбинаторных библиотек вместо фага λ можно использовать нитевидные бактериофаги М13 или fd (рис. 10.14). В этих случаях соответствующий фрагмент антитела синтезируется как часть химерного белка, локализованного на поверхности фаговой частицы. Скрининг комбинаторной библиотеки фрагментов антител можно провести при помощи ферментного иммуносорбентного анализа (ELISA). Суть метода состоит в следующем: образцы (аликвоты) из библиотеки помещают в ячейки планшеты, содержащие антиген-мишень. Ячейки промывают, чтобы удалить несвязанные фаговые частицы. В каждую ячейку вносят конъюгат, состоящий из антитела, связывающегося с белком фаговой оболочки, и фермента. Ячейки промывают для удаления несвязанного конъюгата и добавляют в каждую из них хромогенный субстрат, который расщепляется ферментом, связанным с фагом, и окраши-
220 ГЛАВА 10

|
| Рис. 10.13. Сконструированные участки ДНК из комбинаторной библиотеки кДНК Fv-фрагмента, клонированные в бактериофаг а. А и Б. Фрагменты ДНК L-(A) и Н-(Б) цепей раздельно встроили в векторы на основе бактериофага а. В. Провели рестрикцию каждой EcoRl- библиотеки и лигировали фрагменты ДНК из библиотеки Η-цепи с фрагментами ДНК из библиотеки L-цепи, в результате чего получили комбинаторную библиотеку, содержащую все возможные сочетания фрагментов L- и H -цепей с экспрессией соединенного фрагмента в одном векторе. pLac — laс -промотор Е. соli, ССР - сайт связывания с рибосомой. |
вает те ячейки, в которых находятся фаговые частицы, несущие антитела к антигену-мишени. Процесс отбора и последующая очистка бактериофагов, синтезирующих фрагмент антитела, специфичный к нужному антигену, в этом случае гораздо проще, чем тогда, когда проводится подсчет бляшек бактериофага λ. Выделив фаг, синтезирующий желаемый фрагмент антитела, можно экстрагировать кодирующую этот фрагмент ДНК и субклонировать ее в экспрессирующем векторе. Разные варианты антител с повышенным сродством к антигену-мишени можно получать замещением фрагментов ДНК vl- и Vн-областей или с помощью неспецифического мутагенеза.
Разработав методы получения Fv-фрагментов, исследователи попытались определить, способна ли отдельная белковая цепочка, состоящая только из VL-и Vн-доменов, образовать функциональную молекулу, связывающую антиген. Компьютерное моделирование трехмерной структуры предполагаемого одноцепочечного антитела показало, что для образования конформации, необходимой для связывания антигена, VL- и Vн-домены должны быть разделены линкерным пептидом. Имея это в виду, VL- и VН-ДНК, синтезированные на кДНК-матрице клонированного моноклонального антитела, присоединили к химически синтезированному ДНК-линкеру, создав конструкцию VL-ДНК—линкер—VН-ДНК. Соответствующий одноцепочечный белок синтезировали в Е. coli, очистили и обнаружили, что его сродство и специфичность к антигену сходны с таковыми ин-тактного моноклонального антитела. Таким образом, с помощью Е. coli можно без труда получать функциональные одноцепочечные антитела.
Одноцепочечные антитела могут найти широкое применение в клинике в тех случаях, когда проявление Fc-эффекторных функций не является необходимым, а малый размер молекулы (мол. масса одноцепочечного антитела составляет примерно 27 кДа, а иммуноглобулина G — 150 кДа) дает определенные преимущества. Кроме того, к одноцепочечному антителу можно присоединить последовательность, кодирующую тот или иной белок, получив бифункциональную молекулу, которая сможет связываться с определенной мишенью, проявляя при этом специфическую активность.
Микробиологическое производство лекарственных средств 221
| Рис. 10.14. Создание комбинаторной библиотеки кДНК Fv-фрагментов антител с помощью нитевидного бактериофага М13. кДНК VL- и VH-областей амплифицировали методом ПЦР, а затем лигировали. используя ДНК короткого линкерного пептида. Полученные фрагменты ДНК, составляющие комбинаторную библиотеку кДНК одноцепочечных антител, встроили в геном фага М13 с присоединением их к фаговому гену 3, который кодирует поверхностный фаговый белок. В М13 с гена 3 синтезируется три белковых молекулы, поэтому каждый рекомбинантный фаг M 13, содержащий комбинаторную библиотеку кДНК одноцепочечных антител, будет нести три молекулы химерного белка, состоящего из продуктa гена 3 и одно цепочечного антитела. | 
|
Был проведен также еще один эксперимент: вместо того чтобы соединять VL- и Vн-цепи коротким пептидом, аминокислоты каркасной области модифицировали таким образом, чтобы между ними образовывался дисульфидный мостик. Эффективность такой стабилизированной дисульфидной связью fv-молекулы, связанной с токсином, разрушающим раковые клетки, сравнили с эффективностью одноцепочечной Fv-молекулы, связанной с тем же токсином (рис. 10.15). Обнаружилось, что стабилизированный дисульфидной связью и одноцепочечный Fv-иммунотоксины обладают одинаковой активностью и специфичностью, но первый в несколько раз стабильнее. Можно предположить, что в каких-то ситуациях стабилизированные Fv-молекулы могут оказаться предпочтительнее одноцепочечных Fv-молекул.
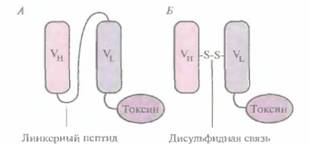
|
| Рис. 10.15. Схематическое изображение одноцепочечного Fv-иммунотоксина (А) и Fv-иммунотоксина, стабилизированного дисульфидной связью (Б). |
222 ГЛАВА 10
Лекарственные средства против ВИЧ
Ученым пока не удалось получить вакцину, достаточно эффективную против вируса иммунодефицита человека (ВИЧ), который вызывает развитие синдрома приобретенного иммунодефицита (СПИД). Параллельно с созданием такой вакцины идет поиск других средств, позволяющих замедлить патологический процесс.
ВИЧ поражает один из видов лимфоцитов, а именно Т-хелперы (Тн-клетки). В норме в процессе развития иммунного ответа Тн-клетки связывают продукты деградации специфических антигенов и высвобождают факторы, стимулирующие другие клетки иммунной системы к участию в иммунном ответе. Тн-клетки играют в этом процессе ключевую роль, а при ВИЧ-инфекции они перестают функционировать. Как только вирус внедряется в Тн-клетку, он становится защищенным от иммунной системы организма и начинает оказывать свое разрушающее действие на Тн-клетки.
• В результате размножения вируса в инфицированной клетке происходит ее лизис.
• Пораженная клетка действует как фабрика по производству ВИЧ-гликопротеина (gpl20), который вызывает разрушение Тн-клеток и других Т-лимфоцитов.
• Пораженная клетка сливается с другими Тн-клетками, формируя синцитий, который не способен выполнять функции, свойственные индивидуальным Тн-клеткам.
Основным следствием ВИЧ инфекции явля ется неспособность иммунной системы организма обеспечивать его защиту от обычных бактериальных и вирусных инфекций, которые в конце концов приводят к гибели больного, несмотря на лечение антибиотиками и другими средствами.
На первом этапе ВИЧ-инфекции происходит взаимодействие между гликопротеином оболочки вируса мол. массой 120 кДа (gpl20) и рецептором на поверхности Тн-клеток — CD4 (рис. 10.l6, A). In vitro поражение th-клеток блокируется антителами к СЕМ; процесс замедляется также при избытке свободного белка CD4. Однако ни один из этих способов не приводит к

|
| Рис. 10.16. ВИЧ-инфекция и ее терапия, А. Связывание ВИЧ с Тн-клеткой опосредуется контактированием вирусного белка gp120 с Тн-клеточным поверхностным белком CD4. Б. На поверхности ВИЧ-инфицированной клетки находится белок gp120, с которым может связываться свободный химерный комплекс С D4— токсин. Попав внутрь инфицированной клетки, токсиновая часть химерной молекулы убивает ее. |
уничтожению вируса. Один из подходов, обеспечивающих как защиту Тн-клеток, так и инактивацию вируса, заключается в создании химерного белка, состоящего из фрагмента молекулы CD4 и Fc-фрагмента иммуноглобулина. Свойства этого белка, называемого СВ4-иммуноад-гезином, определяются составными частями его молекулы: СD4-компонент связывает gp120 и блокирует ВИЧ, а иммуноглобулиновый обеспечивает замедление разрушения молекулы в плазме и ее связывание с клетками, несущими
Микробиологическое производство лекарственных средств 223
рецептор к антителу. После присоединения СD4-иммуноадгезина к свободной вирусной частице или к инфицированной клетке запускается реакция опосредованной антителами клеточной цитотоксичности, которая обеспечивает уничтожение вируса или пораженной им клетки.
Другой подход, позволяющий контролировать развитие ВИЧ-инфекции, заключается в создании системы мечения ВИЧ-пораженных клеток для их специфического уничтожения. Например, если сшить два фрагмента ДНК, один из которых кодирует рецептор CD4, а другой — внутриклеточный токсин Pseitdomonas (экзотоксин А), то мы получим ген, кодирующий химерный белок с комбинированными свойствами (рис. 10.17). Экзотоксин A Pseudomonas -это белок с мол. массой 66 кДа, состоящий из трех доменов: домен I отвечает за связывание с клеткой, II — за проникновение белка в клетку, III — за присоединение ADP-рибозы к эукарио-тическому фактору элонгации (EF-2), что приводит к его инактивации. Химерный белок CD4—экзотоксин A Pseudomonas вместо домена I содержит большую часть последовательности CD4 (рис. 10.17), в результате чего обладает и цитотоксической активностью экзотоксина Pseudomonas, и gp120-связывающей активностью CD4. На поверхности всех ВИЧ-поражен-ных клеток находится гликопротеин gp120, поэтому СD4-домен химерного белка соединяется исключительно с этими клетками. Присоединившись к инфицированной клетке, химерный белок проникает внутрь нее при участии домена II экзотоксина A Pseudomonas. Затем экзотоксиновая часть химерного белка инактивирует фактор элонгации EF-2,участвующий в синтезе белка. Это препятствует дальнейшему синтезу белка, что в конце концов приводит к гибели клетки. Таким образом, CD4-домен «помечает» ВИЧ-пораженные клетки, а экзотоксин выступает в роли «наемного убийцы».
Синтезируясь в Е. coli, химерный белок образует нерастворимые цитоплазматические включения. Их растворяют в гуанидингидрохлориде и выделяют с помощью быстрого разведения и анион-обменной хроматографии. Полученный таким образом белок с успехом выдержал проверку в контрольной культуре клеток. Однако в организме человека на Рsеиdотопаs- компонент химерного белка может возникнуть иммунная реакция, и не исключено, что его придется вводить вместе с каким-либо иммуносупрессантом, например циклоспорином. Нужно иметь в виду, что описанный выше способ борьбы с ВИЧ-инфекцией находится на начальной стадии разработки, хотя в будущем и может оказаться весьма эффективным.
Подобные иммунопрепараты обладают достаточно высокой эффективностью, что позволяет применять их в низких дозах и свести к минимуму побочное действие на иммунную систему. Кроме того, они могут оказаться полезными для лечения различных новообразований, а иногда и заменять химиотерапию. На пораженные клетки можно «нацелить" и другие цитотоксичные белки, например дифтерийный токсин или растительный токсин рицин. Впрочем, даже при
| Рис. 10.17. Генетически сконструированный химерным комплекс СD4— экзотоксин A Pseudomonas. Использован промотор бактериофага Т7 E. coli. | 
|
224 ГЛАВА 10
оптимальном развитии событий пройдет еще несколько лет, прежде чем терапевтическое применение рекомбинантных экзотоксинов станет рутинным.
ЗАКЛЮЧЕНИЕ
С помощью клонирования специфических генов и последующей их экспрессии в бактериях получен целый ряд белков, которые можно будет использовать в качестве лекарственных препаратов. Большинство этих белков имеют эукариотическое происхождение, так что для выделения нужного гена сначала получают препарат мРНК, обогащенный интересующими исследователя фракциями, затем создают кДНК-библиотеку и встраивают соответствующую ДНК в подходящий вектор для экспрессии. Произведя обмен участков родственных генов, кодирующих аналогичные белковые домены, или прямо заменяя сегменты клонированного гена, кодирующие функциональные части белка, можно создавать новые модификации таких белков. В качестве лекарственных средств можно использовать и некоторые ферменты. Например, для снижения вязкости слизи, которая накапливается в легких больных муковисцидозом, применяют в виде аэрозоля рекомбинантную ДНКазу I и альгинатлиазу.
С развитием технологии рекомбинантных ДНК и разработкой способов получения моноклональных антител, а также с установлением структуры и функций иммуноглобулинов появился интерес к использованию специфических антител для лечения различных заболеваний. Работа с генами антител облегчается тем, что отдельные домены молекулы антитела выполняют разные функции.
Лекарственные вещества или ферменты можно присоединять к моноклональным антителам или их Fv-фрагментам, специфичным в отношении поверхностных белков определенных клеток, например опухолевых. При этом лекарственное вещество может находиться в инертной форме. Если предполагаются многократные введения таких комплексов, то их иммуноглобулиновый компонент должен представлять собой антитело или фрагмент антитела человека; это позволяет предотвратить развитие перекрестной иммунной реакции и сенсибилизацию больного. Если же предполагается использовать в этих целях моноклональные антитела грызунов, их структуру следует максимально приблизить к структуре антител человека. Для этого в последних достаточно заменить CDR-участки на аналогичные фрагменты антител грызунов. Недавно удалось провести отбор и синтез моноклональных антител человека с помощью Е. colt.
Генноинженерные методы позволяют получать уникальные лекарственные средства, которые представляют собой комплекс белка, связывающегося со специфическими клетками, например ВИЧ-инфицированными, и токсина. Этот подход пока только разрабатывается, но его перспективы обнадеживают.
ЛИТЕРАТУРА
Barbas C.F., III, D. R. Burton. 1996. Selection and evolution of high-affinity human anti-viral antibodies. Trends Biotechno\. 14: 230-234.
Bird R. E., B. W. Walker. 1991. Single chain antibody variable regions. Trends Btotechnol. 9: 132-137.
Brinkmann U., L. H. Pai, D. J. FitzCerald, M. Wülingham, 1. Pastan. 1991. B3<Fv)-PE38KDEL, a single-chain immunotoxin that causes complete regression of a human carcinoma in mice, Proc. Natl. Acad, Sei. f/&488: 8616-8620.
Brüggemann M., H. M. Caskey, С. Teale, II. Waldmann, G. T. Williams, M. A. Surani, M. S. Mcubcrgcr. 1989. A repertoire of monoclonal antibodies with human heavy chains from transgenic mice. Proc. Nail. Acad. Sei. USA 86: 6709-6713.
Bryn R. A., J. Mordenti, C. Lucas, D. Smith, S. A. Marslers, J. S. Johnson, P. Cossum, S. M. Chamow, F. M. Wurm, T. Gregory, J. E. Gronpman, D. J. Capon. 1990. Biological properties of a CD4 immunoadhesin. Nature 344: 667-670.
Buchner J., R. Rudolph. 1991. Renaturation, purification and characterization of recombinant Fab-fragmnnls produced in E. coli. Bio/Technology 9: 157-162.
Микробиологическое производство лекарственныхсредств 225
Burton D. R. 1991- Human and mouse monoclonal antibodies by repertoire cloning. Trends Biotechnol. 9: 169-175.
Capon D. J., S. M. Chamow, J. Mordent!, S. A. Marsters, T. Gregory, H, Mitsuya, R. A, Bym, С Lucas, F. M. Wurm, J. E. Groopraan, S. Broder, D. H. Smith. 1989. Designing CD4 immunoadhesins for AIDS therapy. Nature 337: 525-530.
Chamow S. M., A. Ashkenazi. 1996. Immunoadhesins: principles and applications. Trends Biotechnol. 14: 52-60.
Chaudhary V, K., T. Mizukami, T. R. Fuerst, D. J. FitzGerald, B. Moss, L Pastan, Έ, A. Berger, 1988. Selective killing of HlV-infected cells by recombinant human CD4-Pseudomonas exotoxin hybrid protein. Nature 335: 369-372.
Chester K. A-, R. E. Hawkins. 1995. Clinical issues in antibody design. Trends Biotechnol. 13: 294—300.
Chiswell D. J., J. McCafferty. 1992. Phage antibodies: will new 'coliclonal' antibodies replace monoclonal antibodies? Trends ßiotechnol. 10: 80-84.
Collet T. A., P. Roben, R. О Kennedy, C. F. Barbas III, D. R. Burton, R. A. Lerner. 1992. A binary plasmid system for shuffling combinatorial antibody libraries. Proc. Natl. Acad. Sei. USA 89: 10026-10030.
Cunningham B. C., J. A. Wells. 1991. Rational design of receptor-specific variants of human growth hormone. Pn>c. Natl. Acad. Set. USA 88: 3407-3411.
Davis G. T., W. D. Bedzyk, E. W. Voss, T. W. Jacobs. 1991. Single chain antibody (SCA) encoding genes: one-step construction and expression in eukaryotic cells, Bio/Technology 9:165-169.
Dewerchin M., D. Collen. 1991. Enhancement of the thrombolytic potency of plasminogen activators by conjugation with clot-specific monoclonal antobodics. Bioconjugaie Chem. 2: 293—300.
Gram H., L. A. Marconi, C. F. Barbas ΙΠ, T. A. Collet, R. A. Lerner, A. S. Kang. 1992. In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc. Natl. Acad. Sei. Î/&489: 3576-3580.
Harris W. J. 1994. Humanizing monoclonal antibodies for in vivo use. Animal Cell Biotechnol. 6: 259-279.
Hodgson,1. 1991, Making monoclonals in microbes. Bio/Technology 9:421^125.
Huennekens F. M. 1994. Tumor targeting: activation of prodrugs by enzymemonoclonal antibody con-jugates. Trends Biotechnol. 12: 234-239.
Huse W. D., L. Sastry, S. A. Iverson, A. S. Kang, M. Alting-Mees, D. R. Burton, S. J. Benkovfc, R. A. Lerner. 1989. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science 246: 1275-1281.
Johnson I. S. 1983, Human insulin from recombinant DNA technology. Science 219: 632-637.
Little M., F. Breitling, S. Dübel, P. Fuchs, M. ßramiage). 1995. Human antibody libraries in Escherichia coli. J. Biotechnol. 41: 187-195.
LoBuglio A. G., R. H. Wheeler, J. Trang, A. Haynes, K. Rogers, E. B. Harvey, L. Sun, J. Ghraycb, M. B. Khazaeli. 1989. Mouse/human chimeric monoclonal antibody in man: kinetics and immune response, Proc. Natl. Acad. Sei. USA 86: 4220-4224.
Marks J. D., A. D. Griffiths, M. Malmqvist, T. P. Oackson, J. M. Bye, G. Winter. 19У2. By-passing immunization: building high affinity antibodies by chain shuffling. Bio/Technology 10: 779-783.
Meyer F., A. Hinnen, A. Meister, M. G. Grutier, S. Alkan. December 1989. Hybrid inlerlcrons. U.S. patent 4, 885,166.
Mullinax R. L., Ε. Λ. Gross, J. R. Amberg, B. N. Hay, H. H. Hogrefe, M. M. Kubitz, A. Greener, M. Alting-Mees, D. Ardourel, J. M. Short, J. A. Sorge, B. Shopes. 1990. identification of human antibody fragment clones specific for tetanus toxoid in a bacteriophage λ immunoex-pression library. Proc. Natl. Acad. Sei. USA 87: 8095-8099.
Murata K., T. Inose, T. Hisano, S. Abe, Y. Yonemnto, T. Yaniashita, M. Takagi, K. Sakaguchi, A. Kimura, T. Imanaka. 1993. Bacterial alginate lyase: enzymology, genetics and appb'cation. /. Ferment. Bioeng. 76:427-437,
Nagata S., H. Taira, A. Hall, L. Johnsrucl, M. Streu«, J. Escodi, W. Boll, K. CanteU, C. Weismann. 1980. Synthesis in E. coli of a polypeptide with human leukocyte interferon activity. Nature 284: 316-320.
Pastan I., D. FitzGerald. 1991. Recombinant toxins for cancer treatment. Science 254: 1173-1177.
Pluckthun A. 1991. Antibody engineering: advances from the use of E. coli expression systems, Bio/Technology 9; 545-551.
226 ГЛАВА 10
Primrose S. B. 1986. The application of genetically engineered microorganisms in the production of drugs. / Appl. Bacterial. 61: 99-116.
Queen C., W. P. Schneider, H. E. Selfck, P. W. Payne, N. F. Landolf, J. F. Duncan, N. M. Avdalovlc, IVL Levitt, R. P. Junghans, T. A. WaWmanih 1989. A humanized antibody that binds to the inter-leukin 2 receptor. Proc. Nail. Acad. Sa. USA 86: 10029-10033.
Reiter Y., U. Brinkmann, К. О. Webber, S.-H. Jung, I. Pastan. 1994. Engineering interchain disulftde bonds into conserved framework regions of Fv fragments: improved biochemical characteristics of recombinant immunotoxins containing disul-fide-stabilized Fv. Protein Eng. 7:697-704.
Riechmann L., M. Clark, H. Waldmann, and G. Winter. 1988. Reshaping human antibodies for therapy. Nature 332: 323-327.
Taniguchi T.t Y. Fiyii-Kuriyama, M. Muramatsu. 1980, Molecular cloning of human Interferon cDNA. Proc. Nat!. Acad. Sei. USA 77:4003-4006.
van Leen R. W., J. G. Bakhuis, R. F. W. С. van Bedthorcn, H. Burger, L· С J. Dorssers, R. W. J. Hommes, P. J. Lemson, B. Noordam, N. L. M. Persoon, G. Wagemaker, 1991. Production of human inter-leukin-3 using industrial microorganisms. Bio/Technology 9: 47-52.
Vaughan T. J., A. J. Williams, K. Pritchard, J. K. Osbourn, A. R. Pope, J. C. Earnshaw, J. M<Caffertv, R. A. Hodits, J. WiKon, K S. Johnson. 19%. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library, Nat. Biotechnol. 14: 309-314.
Waldmann T. A, 1991, Monoclonal antibodies in diagnosis and therapy. Scietice252i 1657—1662.
Winter G., C. Milstein. 1991. Man-made antibodies, Nature 349: 293-299.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Вам нужно клонировать и экспрессировать фрагмент ДНК, кодирующий интерферон человека, У вас нет нужного ДНК-зонда для гибридизации, но вам удалось выделить линию клеток человека, в которых можно индуцировать синтез интерферона с интенсивностью, превышающей фоновую примерно в 100 раз. Какую стратегию клонирования и экспрессии этой ДНК вы выберете?
2. Что такое Fc-фрагмент молекулы антитела? Fab-фрагмент? Fv-фрагмент? CDR-участок?
3. Как осуществляется координированный синтез легкой и тяжелой цепей антитела в Е. соli
4. Какова роль ДНКазы I и альгинатлиазы при лечении муковисцидоза?
5. Как зарегистрировать синтез альгинатлиазы, кодируемой клонированным геном, в трансформированных клетках Е. coif?
6. Что такое комбинаторная библиотека кДНК?
7. Как с помощью бактериофага М13 можно отбирать Fv-фрагменты, связывающиеся со специфическими антигенами-мишенями?
8. Что такое стабилизированная дисульфидными связями и одноцепочечная Fv-молекулы?
9. Как, присоединяя ферменты к моноклональным антителам или их Fv-фрагментам, можно получать лекарственные средства?
10. Как получить моноклональные мышиные антитела, максимально близкие по структуре к антителам человека? Почему они необходимы?
11. Опишите способ получения терапевтического средства, которое «помечает» и уничтожает специфические клетки.
Дата добавления: 2015-07-12; просмотров: 81 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| ГЛАВА 10 | | | СЕРИИ МАШИН ПОСТОЯННОГО ТОКА |