Читайте также:
|
Образование и использование
одноуглеродных фрагментов
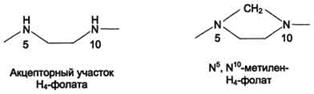
Особое значение реакций катаболизма серина и глицина заключается в том, что они сопровождаются образованием одноуглеродного метиленового фрагмента (-СН2-). Метиленовая группа в молекуле метилен- Н4-фолата может превращаться в другие одноуглеродные группы (фрагменты): метенильную (-СН=), формильную (-НС=О), метильную (-СН3) и формиминогруппу (-CH=NH) (рис. 9-25).
Ещё один источник формального и форми-мино-фрагментов - гистидин. Катаболизм гистидина происходит только в печени (очень небольшой процент в коже) в результате следующих реакций (см. схему на с. 498).
Конечными продуктами катаболизма гистидина являются глутамат, NH3 и одноуглеродные фрагменты - формимино-Н4-фолат и формил-Н4-фолат.
Все образующиеся производные Н4-фолата играют роль промежуточных переносчиков и служат донорами одноуглеродных фрагментов при синтезе некоторых соединений: пуриновых оснований и тимидиловой кислоты (необходимых для синтеза ДНК и РНК), регенерации метионина, синтезе различных формиминопроизводных (формиминоглицина и т.д.)
Перенос одноуглеродных фрагментов к акцептору необходим не только для синтеза ряда соединений, но и для регенерации свободного Н4-фолата в печени.
31. Тетрагидрофолиевая кислота, роль в синтезе и использовании одноуглеродных радикалов. Метилирование гомоцистеина
Ферменты, коферментами которых служат производные фолиевой кислоты играют большую роль в превращениях серина и глицина. Фолиевая кислота – это витамин В9.
- Фолиевая кислота
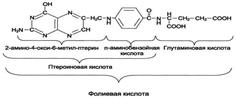
Коферментную функцию выполняет восстановленная форма фолата – тгфк(или Н4-фолат):
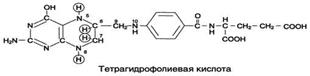
Фолиевая кислота в печени превращается в Н4-фолат в несколько стадий с участием ферментов фолатредуктазы и дигидрофолатредуктазы, коферментом которых служит NADPH.
Особое значение реакций катаболизма серина и глицина заключается в том, что они сопровождаются образованием одноуглеродного метиленового фрагмента (-СН2-), переносчиком которого и является тгфк.
(реакции чисто для наглядности):
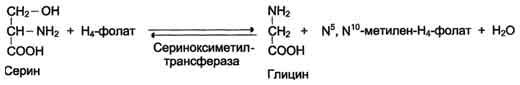
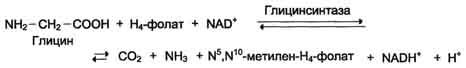
Метиленовая группа в молекуле метилен- Н4-фолата может превращаться в другие одноуглеродные группы (фрагменты): метенильную (-СН=), формильную (-НС=О), метильную (-СН3) и формиминогруппу (-CH=NH)
Таким образом главная роль тгфк - перенос одноуглеродных фрагментов. Они также могут использоваться в дальнейшем для синтеза некоторых соединений: пуриновых оснований и тимидиловой кислоты (необходимых для синтеза ДНК и РНК).
Собственно куда именно присоединяется:
Реакции метилирования играют важную роль в организме и протекают очень интенсивно. Это вызывает большой расход метионина, так как он является незаменимой аминокислотой (в клетках метионин синтезироваться не может). Метионин - незаменимая аминокислота. Активной формой метионина является S-аденозилметионин (SAM) - сульфониевая форма аминокислоты, образующаяся в результате присоединения метионина к молекуле аденозина.
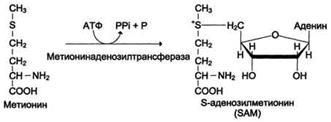
Отщепление метильной группы от SAM и перенос её на соединение-акцептор катализируют ферменты метилтрансферазы. SAM в ходе реакции превращается в S-аденозилгомоцистеин (SAT).
S-аденозилгомоцистеин при действии гидролазы расщепляется на аденозин и гомоцистеин.
S-аденозилгомоцистеин + Н2О → Аденозин + Гомоцистеин
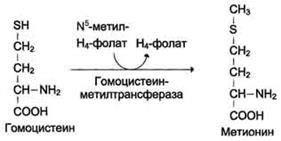
Гомоцистеин может снова превращаться в метионин под действием гомоцистеинметилтранс феразы. Донором метильной группы в этом слу чае служит N5-метил-Н4-фолат.
Метилирование гомоцистеина:
Метионин - незаменимая аминокислота, однако может регенерироваться из гомоцистеина. Следовательно, незаменим именно гомоцистеин, но единственным его источником в организме служит метионин. В пище гомоцистеина крайне мало, поэтому потребности человека в метиони-не и гомоцистеине обеспечиваются только метионином пищи.
Вопрос 32 Недостаточность фолиевой кислоты и витамина В12. Антивитамины фолиевой кислоты. Механизм действия сульфаниламидных препаратов.
Гиповитаминоз фолиевой кислоты приводит к нарушению обмена одноуглеродных фрагментов.
Первое проявление дефицита фолиевой кислоты - мегалобластная (макроцитарная) анемия. Она характеризуется уменьшением количества эритроцитов, снижением содержания в них гемоглобина, что вызывает увеличение размера эритроцитов. Причина этих симптомов - нарушение синтеза ДНК и РНК из-за недостатка их предшественников - тимидиловой кислоты и пуриновых нуклеотидов вследствие дефицита производных Н4-фолата. Клетки кроветворной ткани быстро делятся, поэтому они в первую очередь реагируют на нарушение синтеза нуклеиновых кислот снижением скорости эритропоэза.
Мегалобластная анемия возникает чаще всего в результате недостаточности фолиевой кислоты и/или витамина В12.
Антивитамины фолиевой кислоты:
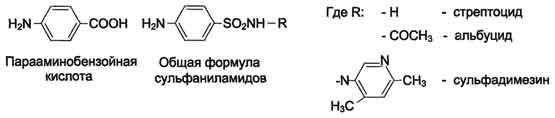
Фолиевая кислота является витамином для человека и животных. Однако многие патогенные бактерии способны синтезировать это соединение, используя парааминобензойную кислоту (ПАБК) - одну из составных частей фолата. ПАБК поступает в бактериальные клетки из внешней среды. Сульфаниламидные лекарственные препараты - производные сульфаниламида (белого стрептоцида), похожи по строению на парааминобензойную кислоту. Отличаются они только радикалами.
Эти препараты подавляют синтез фолиевой кислоты у бактерий, потому что:
конкурентно ингибируют бактериальные ферменты синтеза фолата, так как являются структурными аналогами парааминобензойной кислоты - одного из субстратов процесса;
могут использоваться как псевдосубстраты из-за относительной субстратной специфичности ферментов, в результате чего синтезируется соединение, похожее на фолиевую кислоту, но не выполняющее её функции.
В обоих случаях в клетках бактерий нарушается обмен одноуглеродных фрагментов и, следовательно, синтез нуклеиновых кислот, что вызывает прекращение размножения бактерий.
В клетках больного сульфаниламидные лекарственные вещества не вызывают подобных изменений, поскольку человек получает с пищей готовую фолиевую кислоту.
Вопрос 33.. Обмен фенилаланина и тирозина. Все пути превращения в норме.
Фенилаланин - незаменимая аминокислота, так как в клетках животных не синтезируется её бензольное кольцо. Тирозин - условно заменимая аминокислота, поскольку образуется из фенилаланина.
Основное количество фенилаланина расходуется по 2 путям:
-включается в белки;
-превращается в тирозин.
Превращение фенилаланина в тирозин прежде всего необходимо для удаления избытка фенилаланина, так как высокие концентрации его токсичны для клеток. Образование тирозина не имеет большого значения, так как недостатка этой аминокислоты в клетках практически не бывает.
Основной путь метаболизма фенилаланина начинается с его гидроксилирования (рис. 9-29), в результате чего образуется тирозин. Эта реакция катализируется специфической монооксиге-назой - фенилаланингидроксилазой, коферментом которой служит тетрагидробиоптерин (Н4БП). Активность фермента зависит также от наличия Fe2+. Реакция необратима. Н4БП в результате реакции окисляется в дигидробиоптерин (Н2БП). Регенерация последнего происходит при участии дигидроптеридинредуктазы с использованием NADPH + H+.
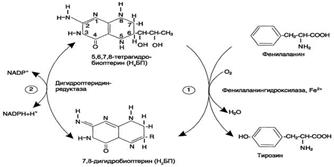
Обмен тирозина значительно сложнее, чем обмен фенилаланина. Кроме использования в синтезе белков, тирозин в разных тканях выступает предшественником таких соединений, как катехоламины, тироксин, меланины, и ка-таболизируется до СО2 и Н2О.
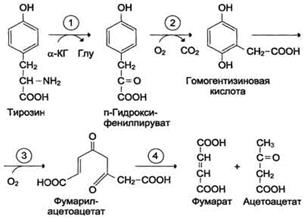
Катаболизм тирозина в печени: Ферменты:
1 реакция – тирозинаминотрансфераза
2 – n-гидроксифенилпируватдиоксигеназа
3-диоксигеназа гомогентизиновой кислоты
4-фумарилацетоацетатгидролаза
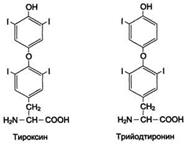
Катаболизм в щитовидной – образование:
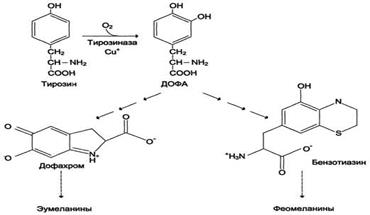
Превращения в меланоцитах – эумеланины и феомеланины. Эумеланины (чёрного и коричневого цвета) - нерастворимые высокомолекулярные гетерополимеры 5,6-дигидроксииндола и некоторых его предшественников. Феомеланины - жёлтые или красновато-коричневые полимеры, растворимые в разбавленных щелочах. Находятся они, в основном, в составе волос. Меланины присутствуют в сетчатке глаз. Цвет кожи зависит от распределения меланоцитов и количества в них разных типов меланинов.Синтез меланинов - сложный, многоступенчатый, разветвлённый процесс. Первую реакцию - превращение тирозина в ДОФА - катализирует тирозиназа, использующая в качестве кофактора ионы Сu+
В надпочечниках:
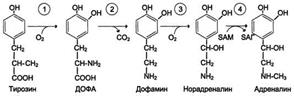
Ферменты: 1-тирозингидроксилаза
2- дофа-декарбоксилаза
3-дофамингидроксидаза
4-метилтрансфераза.
Вопрос 34 - Фенилкетонурия, биохимический дефект, проявление болезни, диагностика, лечение.
В печени здоровых людей небольшая часть фенилаланина (∼10%) превращается в фенил-лактат и фенилацетилглутамин. Этот путь катаболизма фенилаланина становится главным при нарушении основного пути - превращения в тирозин, катализируемого фенилаланингидроксилазой. Такое нарушение сопровождается гиперфенилаланинемией и повышением в крови и моче содержания метаболитов альтернативного пути: фенилпирувата, фенилацетата, фениллактата и фенилацетилглу-тамина.
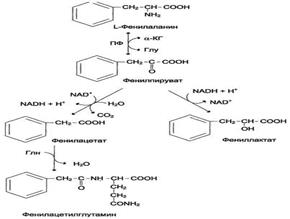
Дефект фенилаланингидроксилазы приводит к заболеванию фенилкетонурия (ФКУ). Выделяют 2 формы ФКУ:
Классическая ФКУ - наследственное заболевание, связанное с мутациями в гене фенилаланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации. При этом концентрация фенилаланина повышается в крови в 20-30 раз
Вариантная ФКУ (коферментзависимая гиперфенилаланинемия) - следствие мутаций в генах, контролирующих метаболизм Н4БП. Клинические проявления - близкие, но не точно совпадающие с проявлениями классической ФКУ. Частота заболевания - 1-2 случая на 1 млн новорождённых.
Прогрессирующее нарушение умственного и физического развития у детей, больных ФКУ, можно предотвратить диетой с очень низким содержанием или полным исключением фенилаланина. Если такое лечение начато сразу после рождения ребёнка, то повреждение мозга предотвращается. Считается, что ограничения в питании могут быть ослаблены после 10-летнего возраста (окончание процессов миелиниза-ции мозга), однако в настоящее время многие педиатры склоняются в сторону "пожизненной диеты".
Для диагностики ФКУ используют качественные и количественные методы обнаружения патологических метаболитов в моче, определение концентрации фенилаланина в крови и моче. Дефектный ген, ответственный за фенилкетонурию, можно обнаружить у фенотипически нормальных гетерозиготных носителей с помощью теста толерантности к фенилаланину. Для этого обследуемому дают натощак ∼10 г фенилаланина в виде раствора, затем через часовые интервалы берут пробы крови, в которых определяют содержание тирозина. В норме концентрация тирозина в крови после фенилаланиновой нагрузки значительно выше, чем у гетерозиготных носителей гена фежилкетонурии. Этот тест используется в генетической консультации для определения риска рождения больного ребёнка. Разработана схема скрининга для выявления новорождённых детей с ФКУ. Чувствительность теста практически достигает 100%.
В настоящее время диагностику мутантного гена, ответственного за ФКУ, можно проводить с помощью методов ДНК-диагностики (рестрикционного анализа и ПЦР).
Вопрос 35. Алкаптонурия, альбинизм. Биохимический дефект, проявление болезней.
Алкаптонурия ("чёрная моча")
Причина заболевания - дефект диоксигеназы гомогентизиновой кислоты. Для этой болезни характерно выделение с мочой большого количества гомогентизиновой кислоты, которая, окисляясь кислородом воздуха, образует тёмные пигменты алкаптоны. Это метаболическое нарушение было описано ещё в XVI веке, а само заболевание охарактеризовано в 1859 г. Клиническими проявлениями болезни, кроме потемнения мочи на воздухе, являются пигментация соединительной ткани (охроноз) и артрит. Частота - 2-5 случаев на 1 млн новорождённых. Заболевание наследуется по аутосомнорецессивному типу. Диагностических методов выявления гетерозиготных носителей дефектного гена к настоящему времени не найдено.
Дата добавления: 2015-09-01; просмотров: 93 | Нарушение авторских прав
| <== предыдущая страница | | | следующая страница ==> |
| Синтез креатина | | | Альбинизм |